

Description
Lujan syndrome is a condition that is characterized by a tall, thin body; distinctive facial features; intellectual disabilities; and weak muscle tone (hypotonia). The condition typically affects men and boys.
After puberty, people with Lujan syndrome are often described as having a marfanoid habitus, because their bodies may resemble those of people with a genetic condition called
Marfan syndrome. For example, people with Lujan syndrome are typically tall and slender and have long fingers and toes with an unusually large range of movement at the joints (hyperextensibility
and toes with an unusually large range of movement at the joints (hyperextensibility ). Although adults with Lujan syndrome are usually tall, their height often falls within the normal range.
). Although adults with Lujan syndrome are usually tall, their height often falls within the normal range.
Additional features seen in people with Lujan syndrome often include an unusually large head (macrocephaly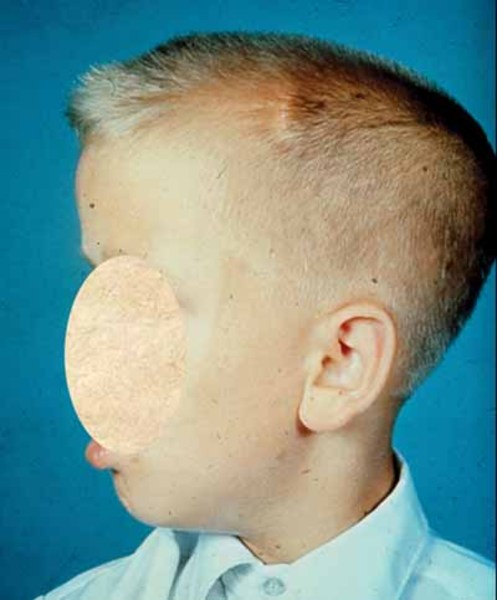 ) and a long, thin face
) and a long, thin face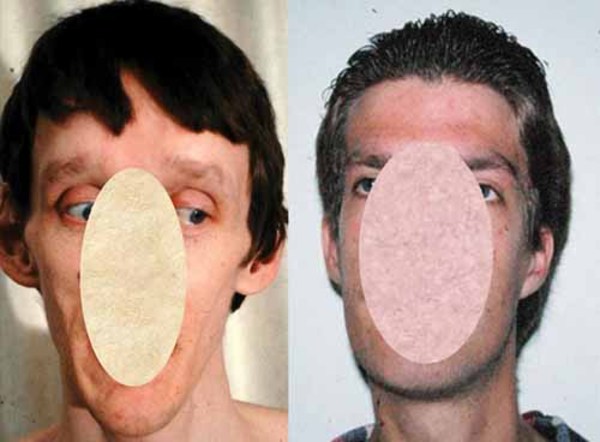 with characteristic features, such as a prominent top of the nose (nasal root); a short space between the nose and the upper lip (philtrum); a narrow roof of the mouth (palate)
with characteristic features, such as a prominent top of the nose (nasal root); a short space between the nose and the upper lip (philtrum); a narrow roof of the mouth (palate) ; crowded teeth
; crowded teeth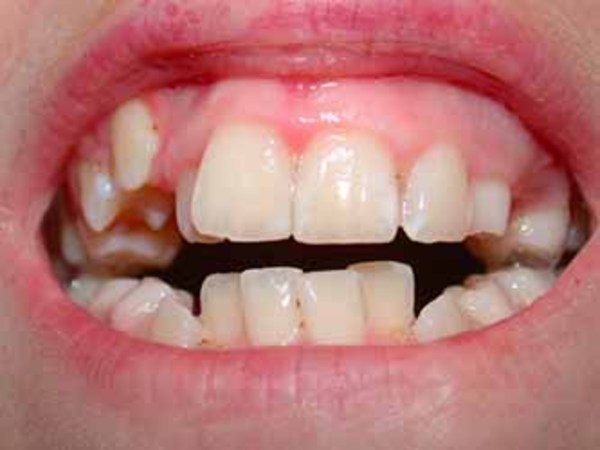 ; and a small chin (micrognathia
; and a small chin (micrognathia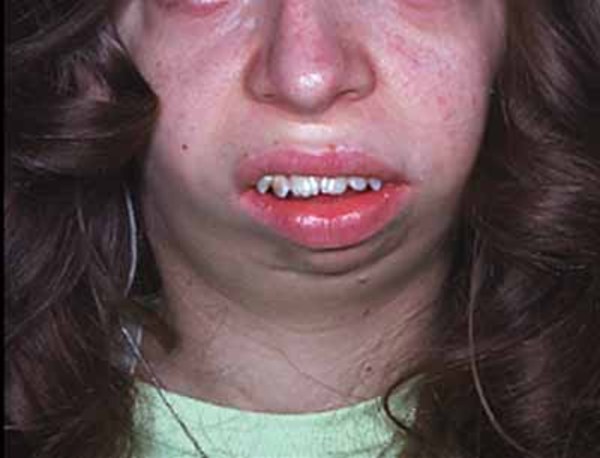 ).
).
Individuals with Lujan syndrome typically have mild to moderate intellectual disabilities. A variety of behavioral problems are common in people with this condition, including hyperactivity, aggressiveness, extreme shyness, or excessive attention-seeking. Some affected individuals have features of autism spectrum disorder or related developmental disorders that affect communication and social interaction. A few people with Lujan syndrome have psychiatric problems, such as delusions and hallucinations.
Additional signs and symptoms of Lujan syndrome can include having an overly nasal voice (hypernasal speech), seizures, abnormalities of the tissue that connects the left and right halves of the brain (corpus callosum), heart defects, and abnormalities of the genitourinary system
(corpus callosum), heart defects, and abnormalities of the genitourinary system .
.
Frequency
Lujan syndrome appears to be very rare, but its exact prevalence is unknown.
Causes
Genetic changes that cause disease are called pathogenic variants. Pathogenic variants in the MED12 gene can cause Lujan syndrome. The MED12 gene provides instructions for making a protein that helps regulate gene activity and is involved in many aspects of early development. Most of the pathogenic variants in the MED12 gene that are associated with Lujan syndrome change a single protein building block (amino acid) in the MED12 protein. This genetic change likely alters the structure and function of the MED12 protein. However, it is unclear exactly how these changes lead to the intellectual disabilities and specific physical features seen in people with Lujan syndrome.
Inheritance
Lujan syndrome is inherited in an X-linked pattern. A condition is considered X-linked if the altered gene that causes the disorder is located on the X chromosome, one of the two sex chromosomes in each cell. In males (who have only one X chromosome), a variant in the only copy of the gene in each cell is typically sufficient to cause the condition. Because they have two X chromosomes, females with a pathogenic variant in one copy of the MED12 gene in each cell are typically not affected. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
in each cell. In males (who have only one X chromosome), a variant in the only copy of the gene in each cell is typically sufficient to cause the condition. Because they have two X chromosomes, females with a pathogenic variant in one copy of the MED12 gene in each cell are typically not affected. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
Other Names for This Condition
- Intellectual disability, X-linked, with marfanoid habitus
- LFS
- Lujan-Fryns syndrome
- X-linked intellectual disability with marfanoid habitus
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Graham JM Jr, Schwartz CE. MED12 related disorders. Am J Med Genet A. 2013 Nov;161A(11):2734-40. doi: 10.1002/ajmg.a.36183. Epub 2013 Oct 10. Citation on PubMed
- Hackmann K, Rump A, Haas SA, Lemke JR, Fryns JP, Tzschach A, Wieczorek D, Albrecht B, Kuechler A, Ripperger T, Kobelt A, Oexle K, Tinschert S, Schrock E, Kalscheuer VM, Di Donato N. Tentative clinical diagnosis of Lujan-Fryns syndrome--A conglomeration of different genetic entities? Am J Med Genet A. 2016 Jan;170A(1):94-102. doi: 10.1002/ajmg.a.37378. Epub 2015 Sep 11. Citation on PubMed
- Lerma-Carrillo I, Molina JD, Cuevas-Duran T, Julve-Correcher C, Espejo-Saavedra JM, Andrade-Rosa C, Lopez-Munoz F. Psychopathology in the Lujan-Fryns syndrome: report of two patients and review. Am J Med Genet A. 2006 Dec 15;140(24):2807-11. doi: 10.1002/ajmg.a.31503. Citation on PubMed
- Lujan JE, Carlin ME, Lubs HA. A form of X-linked mental retardation with marfanoid habitus. Am J Med Genet. 1984 Jan;17(1):311-22. doi: 10.1002/ajmg.1320170124. Citation on PubMed
- Lyons MJ. MED12-Related Disorders. 2008 Jun 23 [updated 2021 Aug 12]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1676/ Citation on PubMed
- Maia N, Ibarluzea N, Misra-Isrie M, Koboldt DC, Marques I, Soares G, Santos R, Marcelis CLM, Keski-Filppula R, Guitart M, Gabau Vila E, Lehman A, Hickey S, Mori M, Terhal P, Valenzuela I, Lasa-Aranzasti A, Cueto-Gonzalez AM, Chhouk BH, Yeh RC, Neil JE, Abu-Libde B, Kleefstra T, Elting MW, Csaszar A, Karteszi J, Bessenyei B, van Bokhoven H, Jorge P, van Hagen JM, de Brouwer APM. Missense MED12 variants in 22 males with intellectual disability: From nonspecific symptoms to complete syndromes. Am J Med Genet A. 2023 Jan;191(1):135-143. doi: 10.1002/ajmg.a.63004. Epub 2022 Oct 22. Citation on PubMed
- Schwartz CE, Tarpey PS, Lubs HA, Verloes A, May MM, Risheg H, Friez MJ, Futreal PA, Edkins S, Teague J, Briault S, Skinner C, Bauer-Carlin A, Simensen RJ, Joseph SM, Jones JR, Gecz J, Stratton MR, Raymond FL, Stevenson RE. The original Lujan syndrome family has a novel missense mutation (p.N1007S) in the MED12 gene. J Med Genet. 2007 Jul;44(7):472-7. doi: 10.1136/jmg.2006.048637. Epub 2007 Mar 16. Citation on PubMed or Free article on PubMed Central
- Van Buggenhout G, Fryns JP. Lujan-Fryns syndrome (mental retardation, X-linked, marfanoid habitus). Orphanet J Rare Dis. 2006 Jul 10;1:26. doi: 10.1186/1750-1172-1-26. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.