

Description
Knobloch syndrome is a rare condition characterized by severe vision problems and a skull defect.
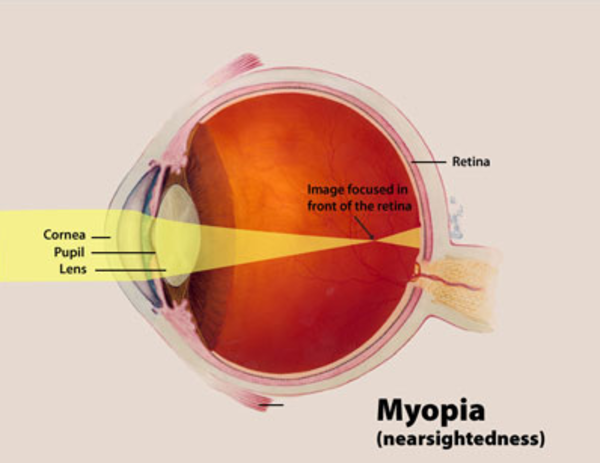
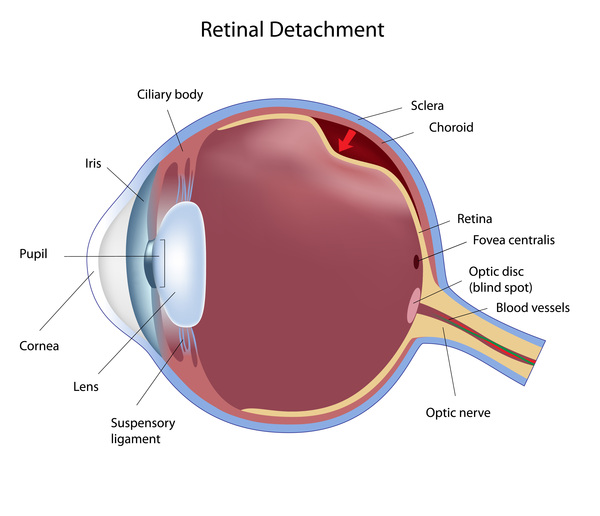
A characteristic feature of Knobloch syndrome is extreme nearsightedness (high myopia). In addition, several other eye abnormalities are common in people with this condition. Most affected individuals have vitreoretinal degeneration, which is breakdown (degeneration) of two structures in the eye called the vitreous and the retina. The vitreous is the gelatin-like substance that fills the eye, and the retina is the light-sensitive tissue at the back of the eye. Vitreoretinal degeneration often leads to separation of the retina from the back of the eye (retinal detachment). Affected individuals may also have abnormalities in the central area of the retina, called the macula. The macula is responsible for sharp central vision, which is needed for detailed tasks such as reading, driving, and recognizing faces. Due to abnormalities in the vitreous, retina, and macula, people with Knobloch syndrome often develop blindness in one or both eyes.
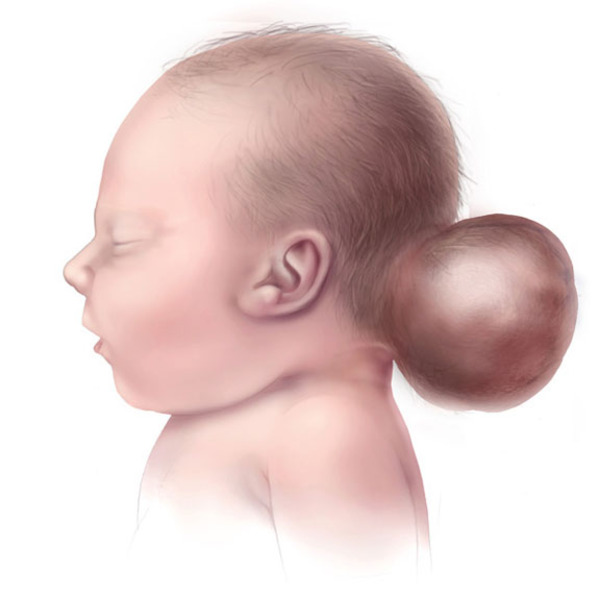
Another characteristic feature of Knobloch syndrome is a skull defect called an occipital encephalocele, which is a sac-like protrusion of the brain (encephalocele) through a defect in the bone at the base of the skull (occipital bone). Some affected individuals have been diagnosed with a different skull defect in the occipital region, and it is unclear whether the defect is always a true encephalocele. In other conditions, encephaloceles may be associated with intellectual disability; however, most people with Knobloch syndrome have normal intelligence.
Frequency
Knobloch syndrome is a rare condition. However, the exact prevalence of the condition is unknown.
Causes
Mutations in the COL18A1 gene can cause Knobloch syndrome. The COL18A1 gene provides instructions for making a protein that forms collagen XVIII, which is found in the basement membranes of tissues throughout the body. Basement membranes are thin, sheet-like structures that separate and support cells in these tissues. Collagen XVIII is found in the basement membranes of several parts of the eye, including the vitreous and retina, among other tissues. Little is known about the function of this protein, but it appears to be involved in normal development of the eye.
Several mutations in the COL18A1 gene have been identified in people with Knobloch syndrome. Most COL18A1 gene mutations lead to an abnormally short version of the genetic blueprint used to make the collagen XVIII protein. Although the process is unclear, the COL18A1 gene mutations result in the loss of collagen XVIII protein, which likely causes the signs and symptoms of Knobloch syndrome.
When the condition is caused by COL18A1 gene mutations, it is sometimes referred to as Knobloch syndrome type I. Research indicates that mutations in at least two other genes that have not been identified may cause Knobloch syndrome types II and III. Although they are caused by alterations in different genes, the three types of the condition have similar signs and symptoms.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Retinal detachment and occipital encephalocele
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Fukai N, Eklund L, Marneros AG, Oh SP, Keene DR, Tamarkin L, Niemela M, Ilves M, Li E, Pihlajaniemi T, Olsen BR. Lack of collagen XVIII/endostatin results in eye abnormalities. EMBO J. 2002 Apr 2;21(7):1535-44. doi: 10.1093/emboj/21.7.1535. Citation on PubMed or Free article on PubMed Central
- Passos-Bueno MR, Suzuki OT, Armelin-Correa LM, Sertie AL, Errera FI, Bagatini K, Kok F, Leite KR. Mutations in collagen 18A1 and their relevance to the human phenotype. An Acad Bras Cienc. 2006 Mar;78(1):123-31. doi: 10.1590/s0001-37652006000100012. Epub 2006 Mar 8. Citation on PubMed
- Seaver LH, Joffe L, Spark RP, Smith BL, Hoyme HE. Congenital scalp defects and vitreoretinal degeneration: redefining the Knobloch syndrome. Am J Med Genet. 1993 Apr 15;46(2):203-8. doi: 10.1002/ajmg.1320460221. Citation on PubMed
- Seppinen L, Pihlajaniemi T. The multiple functions of collagen XVIII in development and disease. Matrix Biol. 2011 Mar;30(2):83-92. doi: 10.1016/j.matbio.2010.11.001. Epub 2010 Dec 14. Citation on PubMed
- Sertie AL, Sossi V, Camargo AA, Zatz M, Brahe C, Passos-Bueno MR. Collagen XVIII, containing an endogenous inhibitor of angiogenesis and tumor growth, plays a critical role in the maintenance of retinal structure and in neural tube closure (Knobloch syndrome). Hum Mol Genet. 2000 Aug 12;9(13):2051-8. doi: 10.1093/hmg/9.13.2051. Citation on PubMed
- Suzuki O, Kague E, Bagatini K, Tu H, Heljasvaara R, Carvalhaes L, Gava E, de Oliveira G, Godoi P, Oliva G, Kitten G, Pihlajaniemi T, Passos-Bueno MR. Novel pathogenic mutations and skin biopsy analysis in Knobloch syndrome. Mol Vis. 2009;15:801-9. Epub 2009 Apr 23. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.