

Description
Kearns-Sayre syndrome is a condition that affects many parts of the body, especially the eyes. The features of Kearns-Sayre syndrome usually appear before age 20, and the condition is diagnosed by a few characteristic signs and symptoms. People with Kearns-Sayre syndrome have progressive external ophthalmoplegia, which is weakness or paralysis of the eye muscles that impairs eye movement and causes drooping eyelids (ptosis). Affected individuals also have an eye condition called pigmentary retinopathy, which results from breakdown (degeneration) of the light-sensing tissue at the back of the eye (the retina) that gives it a speckled and streaked appearance. The retinopathy may cause loss of vision. In addition, people with Kearns-Sayre syndrome have at least one of the following signs or symptoms: abnormalities of the electrical signals that control the heartbeat (cardiac conduction defects), problems with coordination and balance that cause unsteadiness while walking (ataxia), or abnormally high levels of protein in the fluid that surrounds and protects the brain and spinal cord (the cerebrospinal fluid or CSF).
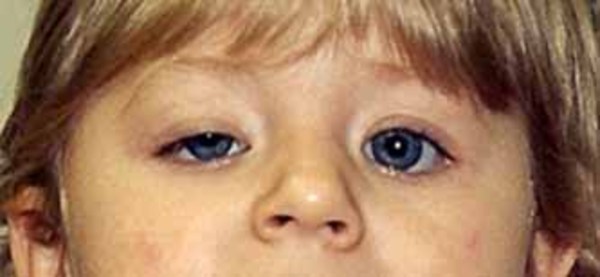
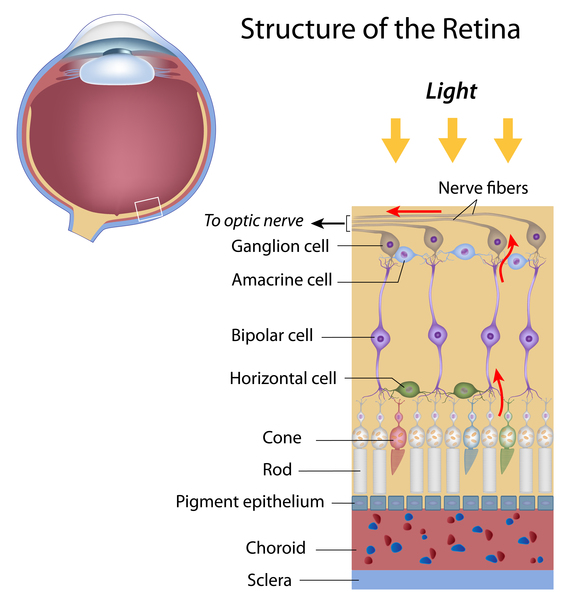
People with Kearns-Sayre syndrome may also experience muscle weakness in their limbs, deafness, kidney problems, or a deterioration of cognitive functions (dementia). Affected individuals often have short stature. In addition, diabetes mellitus is occasionally seen in people with Kearns-Sayre syndrome.
When the muscle cells of affected individuals are stained and viewed under a microscope, these cells usually appear abnormal. The abnormal muscle cells contain an excess of structures called mitochondria and are known as ragged-red fibers.
A related condition called ophthalmoplegia-plus may be diagnosed if an individual has many of the signs and symptoms of Kearns-Sayre syndrome but not all the criteria are met.
Frequency
The prevalence of Kearns-Sayre syndrome is approximately 1 to 3 per 100,000 individuals.
Causes
Kearns-Sayre syndrome is a condition caused by defects in mitochondria, which are structures within cells that use oxygen to convert the energy from food into a form cells can use. This process is called oxidative phosphorylation. Although most DNA is packaged in chromosomes within the nucleus (nuclear DNA), mitochondria also have a small amount of their own DNA, called mitochondrial DNA (mtDNA). This type of DNA contains many genes essential for normal mitochondrial function. People with Kearns-Sayre syndrome have a single, large deletion of mtDNA, ranging from 1,000 to 10,000 DNA building blocks (nucleotides). The cause of the deletion in affected individuals is unknown.

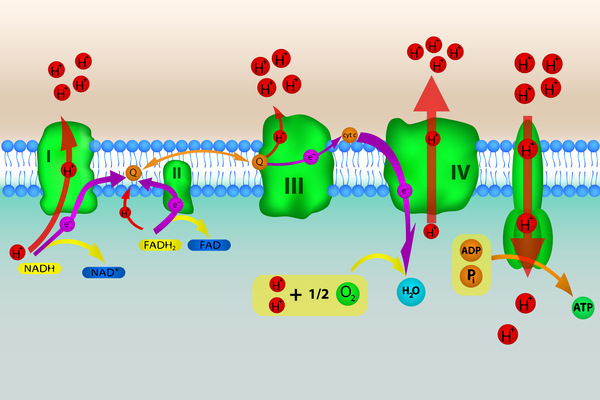
The mtDNA deletions that cause Kearns-Sayre syndrome result in the loss of genes important for mitochondrial protein formation and oxidative phosphorylation. The most common deletion removes 4,997 nucleotides, which includes twelve mitochondrial genes. Deletions of mtDNA result in impairment of oxidative phosphorylation and a decrease in cellular energy production. Regardless of which genes are deleted, all steps of oxidative phosphorylation are affected. Researchers have not determined how these deletions lead to the specific signs and symptoms of Kearns-Sayre syndrome, although the features of the condition are probably related to a lack of cellular energy. It has been suggested that eyes are commonly affected by mitochondrial defects because they are especially dependent on mitochondria for energy.
Inheritance
This condition is generally not inherited but arises from mutations in the body's cells that occur after conception. This alteration is called a somatic mutation and is present only in certain cells.
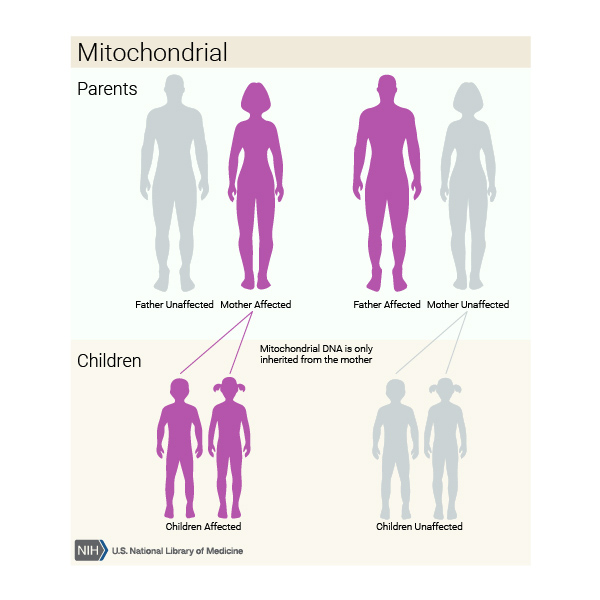
Rarely, this condition is inherited in a mitochondrial pattern, which is also known as maternal inheritance. This pattern of inheritance applies to genes contained in mtDNA. Because egg cells, but not sperm cells, contribute mitochondria to the developing embryo, children can only inherit disorders resulting from mtDNA mutations from their mother. These disorders can appear in every generation of a family and can affect both males and females, but fathers do not pass traits associated with changes in mtDNA to their children.
Other Names for This Condition
- Kearns-Sayre mitochondrial cytopathy
- KSS
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Goldstein A, Falk MJ. Single Large-Scale Mitochondrial DNA Deletion Syndromes. 2003 Dec 17 [updated 2023 Sep 28]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1203/ Citation on PubMed
- Graeber MB, Muller U. Recent developments in the molecular genetics of mitochondrial disorders. J Neurol Sci. 1998 Jan 8;153(2):251-63. doi: 10.1016/s0022-510x(97)00295-5. Citation on PubMed
- Porteous WK, James AM, Sheard PW, Porteous CM, Packer MA, Hyslop SJ, Melton JV, Pang CY, Wei YH, Murphy MP. Bioenergetic consequences of accumulating the common 4977-bp mitochondrial DNA deletion. Eur J Biochem. 1998 Oct 1;257(1):192-201. doi: 10.1046/j.1432-1327.1998.2570192.x. Citation on PubMed
- Schroder R, Vielhaber S, Wiedemann FR, Kornblum C, Papassotiropoulos A, Broich P, Zierz S, Elger CE, Reichmann H, Seibel P, Klockgether T, Kunz WS. New insights into the metabolic consequences of large-scale mtDNA deletions: a quantitative analysis of biochemical, morphological, and genetic findings in human skeletal muscle. J Neuropathol Exp Neurol. 2000 May;59(5):353-60. doi: 10.1093/jnen/59.5.353. Citation on PubMed
- Sciacco M, Bonilla E, Schon EA, DiMauro S, Moraes CT. Distribution of wild-type and common deletion forms of mtDNA in normal and respiration-deficient muscle fibers from patients with mitochondrial myopathy. Hum Mol Genet. 1994 Jan;3(1):13-9. doi: 10.1093/hmg/3.1.13. Citation on PubMed
- Yamashita S, Nishino I, Nonaka I, Goto YI. Genotype and phenotype analyses in 136 patients with single large-scale mitochondrial DNA deletions. J Hum Genet. 2008;53(7):598. doi: 10.1007/s10038-008-0289-8. Epub 2008 Apr 15. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.