

Description
Juvenile Paget disease is a disorder that affects bone growth. This disease causes bones to be abnormally large, misshapen, and easily broken (fractured). The specific signs and symptoms and the severity of the condition can vary among affected individuals.
The features of juvenile Paget disease appear in infancy or childhood. As bones grow, they become weaker and misshapen. These abnormalities usually become more severe during the adolescent growth spurt, when bones grow very quickly.
Juvenile Paget disease affects the entire skeleton, resulting in widespread bone and joint pain. The bones of the skull tend to grow unusually large and thick, which can increase the size of the head (circumference). The abnormal growth of the skull bones can damage the bones in the ear, leading to hearing loss. The disease can also cause an abnormal curvature of the spine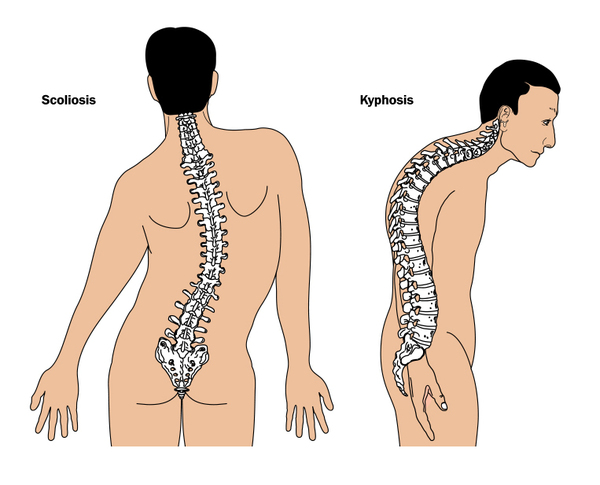 (kyphosis). Additionally, the weight-bearing long bones in the legs tend to bow and fracture easily, which can interfere with the ability to stand and walk.
(kyphosis). Additionally, the weight-bearing long bones in the legs tend to bow and fracture easily, which can interfere with the ability to stand and walk.
Other features of juvenile Paget disease can include short stature; developmental delays; dental problems, such as the delayed appearance (eruption) of teeth or the early (premature) loss of teeth; and vision problems, which can include abnormalities of the light-sensing tissue at the back of the eye (retina). Affected individuals may also have an abnormal accumulation of calcium (calcification) in the walls of blood vessels. Rarely, people with juvenile Paget disease have a bulge (aneurysm) in the wall of the vessel that carries blood to the brain, face, and neck (internal carotid artery). If an aneurysm grows large, it can burst and cause dangerous bleeding.
Frequency
Juvenile Paget disease is extremely rare; approximately 100 affected individuals have been reported in the medical literature.
Causes
Variants (also called mutations) in the TNFRSF11B gene cause juvenile Paget disease. This gene provides instructions for making a protein that is involved in bone remodeling, a normal process in which old bone is broken down and replaced by new bone. Bones are constantly being remodeled, and the process is carefully controlled to ensure that bones stay strong and healthy.
The variants in the TNFRSF11B gene that are associated with juvenile Paget disease cause cells to produce a version of the protein that does not function properly. As a result, bone is broken down and then replaced at a faster rate than usual. The new bone tissue is larger, less organized, and weaker than normal bone. This abnormally fast bone remodeling leads to the features seen in people with juvenile Paget disease.
Inheritance
Juvenile Paget disease is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
, which means both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
Other Names for This Condition
- Chronic congenital idiopathic hyperphosphatasemia
- Familial Hyperphosphatasemia
- Familial hyperphosphatasia
- Familial idiopathic hyperphosphatasia
- Familial osteoectasia
- Hereditary hyperphosphatasia
- Hyperostosis corticalis deformans juvenilis
- Idiopathic hyperphosphatasia
- JPD
- Paget disease of bone 5
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Chong B, Hegde M, Fawkner M, Simonet S, Cassinelli H, Coker M, Kanis J, Seidel J, Tau C, Tuysuz B, Yuksel B, Love D; International Hyperphosphatasia Collaborative Group. Idiopathic hyperphosphatasia and TNFRSF11B mutations: relationships between phenotype and genotype. J Bone Miner Res. 2003 Dec;18(12):2095-104. doi: 10.1359/jbmr.2003.18.12.2095. Citation on PubMed
- Cundy T, Hegde M, Naot D, Chong B, King A, Wallace R, Mulley J, Love DR, Seidel J, Fawkner M, Banovic T, Callon KE, Grey AB, Reid IR, Middleton-Hardie CA, Cornish J. A mutation in the gene TNFRSF11B encoding osteoprotegerin causes an idiopathic hyperphosphatasia phenotype. Hum Mol Genet. 2002 Sep 1;11(18):2119-27. doi: 10.1093/hmg/11.18.2119. Citation on PubMed
- Janssens K, de Vernejoul MC, de Freitas F, Vanhoenacker F, Van Hul W. An intermediate form of juvenile Paget's disease caused by a truncating TNFRSF11B mutation. Bone. 2005 Mar;36(3):542-8. doi: 10.1016/j.bone.2004.12.004. Citation on PubMed
- Lucas GJ, Daroszewska A, Ralston SH. Contribution of genetic factors to the pathogenesis of Paget's disease of bone and related disorders. J Bone Miner Res. 2006 Dec;21 Suppl 2:P31-7. doi: 10.1359/jbmr.06s206. Citation on PubMed
- Naot D, Choi A, Musson DS, Simsek Kiper PO, Utine GE, Boduroglu K, Peacock M, DiMeglio LA, Cundy T. Novel homozygous mutations in the osteoprotegerin gene TNFRSF11B in two unrelated patients with juvenile Paget's disease. Bone. 2014 Nov;68:6-10. doi: 10.1016/j.bone.2014.07.034. Epub 2014 Aug 6. Citation on PubMed
- Polyzos SA, Cundy T, Mantzoros CS. Juvenile Paget disease. Metabolism. 2018 Mar;80:15-26. doi: 10.1016/j.metabol.2017.10.007. Epub 2017 Nov 22. Citation on PubMed
- Ralston SH, Taylor JP. Rare Inherited forms of Paget's Disease and Related Syndromes. Calcif Tissue Int. 2019 May;104(5):501-516. doi: 10.1007/s00223-019-00520-5. Epub 2019 Feb 13. Citation on PubMed
- Ralston SH. Juvenile Paget's disease, familial expansile osteolysis and other genetic osteolytic disorders. Best Pract Res Clin Rheumatol. 2008 Mar;22(1):101-11. doi: 10.1016/j.berh.2007.11.005. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.