

Description
Junctional epidermolysis bullosa (JEB) is a major form of epidermolysis bullosa, a group of genetic conditions that cause the skin to be very fragile and to blister easily. Blisters and areas of skin loss (erosions) form in response to minor injury or friction, such as rubbing or scratching. Researchers classify junctional epidermolysis bullosa into two main types: JEB generalized severe (formerly known as Herlitz JEB) and JEB generalized intermediate (formerly known as non-Herlitz JEB). Although the types differ in severity, their features overlap significantly, and they can be caused by mutations in the same genes.
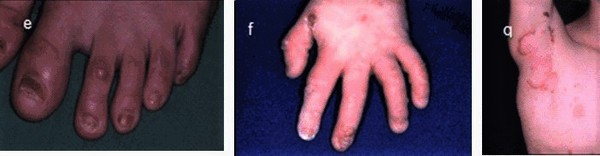
JEB generalized severe is the more serious form of the condition. From birth or early infancy, affected individuals have blistering over large regions of the body. Blistering also affects the mucous membranes, such as the moist lining of the mouth and digestive tract, which can make it difficult to eat and digest food. As a result, many affected children are undernourished and grow slowly. The extensive blistering leads to scarring and the formation of red, bumpy patches called granulation tissue. Granulation tissue bleeds easily and profusely, making affected infants susceptible to serious infections and loss of necessary proteins, minerals, and fluids. Additionally, a buildup of granulation tissue in the airway can lead to a weak, hoarse cry and difficulty breathing.
Other complications of JEB generalized severe can include fusion of the fingers and toes, abnormalities of the fingernails and toenails, joint deformities (contractures) that limit movement, hair loss (alopecia), and thinning of the protective outer layer (enamel) of the teeth. Because the signs and symptoms of JEB generalized severe are so serious, infants with this condition usually do not survive beyond the first year of life.
The milder form of junctional epidermolysis bullosa is called JEB generalized intermediate. The blistering associated with JEB generalized intermediate may be limited to the hands, feet, knees, and elbows, and it often improves after the newborn period. Other characteristic features of this form of the condition include hair loss, abnormal fingernails and toenails, and irregular tooth enamel. Most affected individuals do not have extensive scarring or granulation tissue formation, so breathing difficulties and other severe complications are rare. JEB generalized intermediate is typically associated with a normal lifespan.
Frequency
Both types of junctional epidermolysis bullosa are rare, together affecting approximately 3 per million people per year in the United States.
Causes
Junctional epidermolysis bullosa most commonly results from mutations in the LAMA3, LAMB3, LAMC2, and COL17A1 genes. Mutations in each of these genes can cause JEB generalized severe or JEB generalized intermediate. LAMB3 gene mutations are the most common, causing about 70 percent of all cases of junctional epidermolysis bullosa.
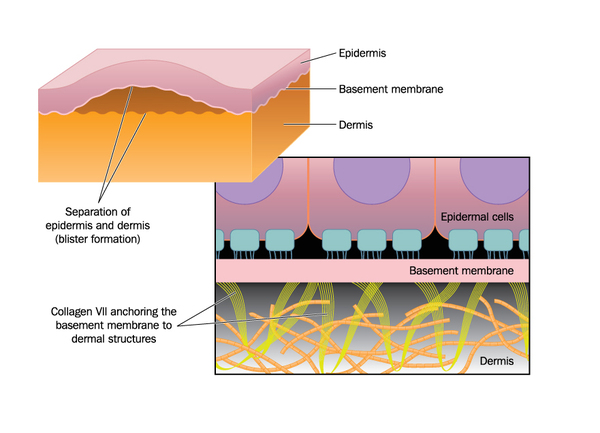
The LAMA3, LAMB3, and LAMC2 genes each provide instructions for making one part (subunit) of a protein called laminin 332. This protein plays an important role in strengthening and stabilizing the skin by helping to attach the top layer of skin (the epidermis) to underlying layers. Mutations in any of the three laminin 332 genes lead to the production of a defective or nonfunctional version of this protein. Without functional laminin 332, cells in the epidermis are fragile and easily damaged. Friction or other minor trauma can cause the skin layers to separate, leading to the formation of blisters.
The COL17A1 gene provides instructions for making a protein that is used to assemble type XVII collagen. Collagens are molecules that give structure and strength to connective tissues, such as skin, tendons, and ligaments, throughout the body. Type XVII collagen helps attach the epidermis to underlying layers of skin, making the skin strong and flexible. Mutations in the COL17A1 gene prevent the normal formation of collagen XVII. As a result, the skin is less resistant to friction and minor trauma and blisters easily. Most COL17A1 gene mutations cause JEB generalized intermediate, although a few individuals with mutations in this gene have had the more serious JEB generalized severe.
Very rarely, junctional epidermolysis bullosa is caused by mutations in another gene that provides instructions for making a different protein that helps attach the top layer of skin to underlying layers.
Inheritance
Both types of junctional epidermolysis bullosa are inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. Rarely, people with one mutated copy of the COL17A1, LAMA3, or LAMB3 gene have irregular tooth enamel.

Other Names for This Condition
- Epidermolysis bullosa, junctional
- JEB
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Castori M, Floriddia G, De Luca N, Pascucci M, Ghirri P, Boccaletti V, El Hachem M, Zambruno G, Castiglia D. Herlitz junctional epidermolysis bullosa: laminin-5 mutational profile and carrier frequency in the Italian population. Br J Dermatol. 2008 Jan;158(1):38-44. doi: 10.1111/j.1365-2133.2007.08208.x. Epub 2007 Oct 4. Citation on PubMed
- Condrat I, He Y, Cosgarea R, Has C. Junctional Epidermolysis Bullosa: Allelic Heterogeneity and Mutation Stratification for Precision Medicine. Front Med (Lausanne). 2019 Jan 29;5:363. doi: 10.3389/fmed.2018.00363. eCollection 2018. Citation on PubMed or Free article on PubMed Central
- Fine JD, Eady RA, Bauer EA, Bauer JW, Bruckner-Tuderman L, Heagerty A, Hintner H, Hovnanian A, Jonkman MF, Leigh I, McGrath JA, Mellerio JE, Murrell DF, Shimizu H, Uitto J, Vahlquist A, Woodley D, Zambruno G. The classification of inherited epidermolysis bullosa (EB): Report of the Third International Consensus Meeting on Diagnosis and Classification of EB. J Am Acad Dermatol. 2008 Jun;58(6):931-50. doi: 10.1016/j.jaad.2008.02.004. Epub 2008 Apr 18. Citation on PubMed
- Has C, Liu L, Bolling MC, Charlesworth AV, El Hachem M, Escamez MJ, Fuentes I, Buchel S, Hiremagalore R, Pohla-Gubo G, van den Akker PC, Wertheim-Tysarowska K, Zambruno G. Clinical practice guidelines for laboratory diagnosis of epidermolysis bullosa. Br J Dermatol. 2020 Mar;182(3):574-592. doi: 10.1111/bjd.18128. Epub 2019 Aug 9. Citation on PubMed or Free article on PubMed Central
- Kelly-Mancuso G, Kopelan B, Azizkhan RG, Lucky AW. Junctional epidermolysis bullosa incidence and survival: 5-year experience of the Dystrophic Epidermolysis Bullosa Research Association of America (DebRA) nurse educator, 2007 to 2011. Pediatr Dermatol. 2014 Mar-Apr;31(2):159-62. doi: 10.1111/pde.12157. Epub 2013 May 31. Citation on PubMed
- Muhle C, Jiang QJ, Charlesworth A, Bruckner-Tuderman L, Meneguzzi G, Schneider H. Novel and recurrent mutations in the laminin-5 genes causing lethal junctional epidermolysis bullosa: molecular basis and clinical course of Herlitz disease. Hum Genet. 2005 Jan;116(1-2):33-42. doi: 10.1007/s00439-004-1210-y. Epub 2004 Nov 5. Citation on PubMed
- Nakano A, Lestringant GG, Paperna T, Bergman R, Gershoni R, Frossard P, Kanaan M, Meneguzzi G, Richard G, Pfendner E, Uitto J, Pulkkinen L, Sprecher E. Junctional epidermolysis bullosa in the Middle East: clinical and genetic studies in a series of consanguineous families. J Am Acad Dermatol. 2002 Apr;46(4):510-6. doi: 10.1067/mjd.2002.119673. Citation on PubMed
- Pfendner EG, Bruckner A, Conget P, Mellerio J, Palisson F, Lucky AW. Basic science of epidermolysis bullosa and diagnostic and molecular characterization: Proceedings of the IInd International Symposium on Epidermolysis Bullosa, Santiago, Chile, 2005. Int J Dermatol. 2007 Aug;46(8):781-94. doi: 10.1111/j.1365-4632.2007.03307.x. No abstract available. Citation on PubMed
- Pfendner EG, Lucky AW. Junctional Epidermolysis Bullosa. 2008 Feb 22 [updated 2018 Dec 20]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1125/ Citation on PubMed
- Pulkkinen L, Uitto J. Mutation analysis and molecular genetics of epidermolysis bullosa. Matrix Biol. 1999 Feb;18(1):29-42. doi: 10.1016/s0945-053x(98)00005-5. Citation on PubMed
- Varki R, Sadowski S, Pfendner E, Uitto J. Epidermolysis bullosa. I. Molecular genetics of the junctional and hemidesmosomal variants. J Med Genet. 2006 Aug;43(8):641-52. doi: 10.1136/jmg.2005.039685. Epub 2006 Feb 10. Citation on PubMed or Free article on PubMed Central
- Yoshida K, Sadamoto M, Sasaki T, Kubo A, Ishiko A. Junctional epidermolysis bullosa without pyloric atresia due to a homozygous missense mutation in ITGB4. J Dermatol. 2019 Feb;46(2):e61-e63. doi: 10.1111/1346-8138.14591. Epub 2018 Aug 6. No abstract available. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.