

Description
Incontinentia pigmenti is a condition that can affect many body systems, particularly the skin . This condition occurs much more often in females than in males.
. This condition occurs much more often in females than in males.
Incontinentia pigmenti is characterized by skin abnormalities that typically evolve throughout childhood and young adulthood. Many affected infants have a blistering rash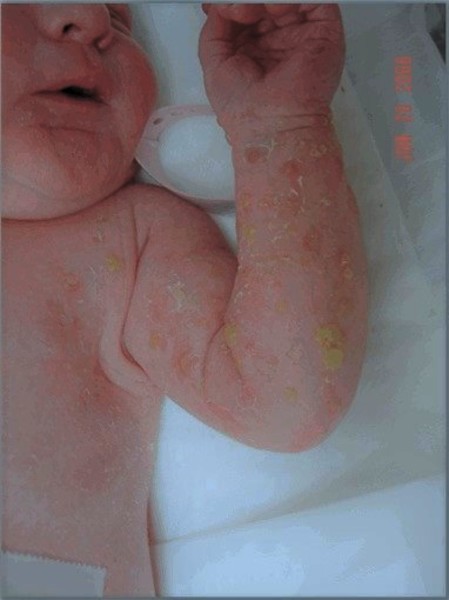 at birth and in early infancy. Though this blistering heals spontaneously, it can recur during illnesses with high fever. This blistering stage is followed by the development of wart-like (verrucous) lesions
at birth and in early infancy. Though this blistering heals spontaneously, it can recur during illnesses with high fever. This blistering stage is followed by the development of wart-like (verrucous) lesions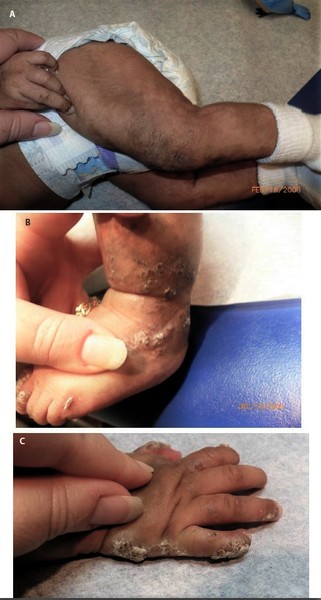 that also heal spontaneously. The blisters and wart-like lesions primarily occur on the arms and legs.
that also heal spontaneously. The blisters and wart-like lesions primarily occur on the arms and legs.
In infancy and early childhood, the skin develops grey or brown patches (hyperpigmentation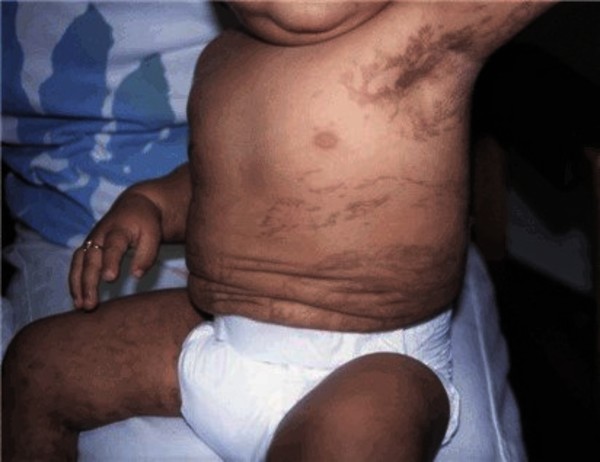 ) that occur in a swirled pattern. These patches, which can occur anywhere on the body, fade with time. Adults with incontinentia pigmenti usually have lines of unusually light-colored skin (hypopigmentation) on their arms and legs. These markings follow the paths along which cells migrate as the skin develops before birth (called the lines of Blaschko).
) that occur in a swirled pattern. These patches, which can occur anywhere on the body, fade with time. Adults with incontinentia pigmenti usually have lines of unusually light-colored skin (hypopigmentation) on their arms and legs. These markings follow the paths along which cells migrate as the skin develops before birth (called the lines of Blaschko).
Individuals with incontinentia pigmenti are at risk of stroke and vision loss, especially within the first year of life. These risks are due to abnormalities in blood vessels in the brain and in the light-sensitive tissue that lines the back of the eye
(retina
and vision loss, especially within the first year of life. These risks are due to abnormalities in blood vessels in the brain and in the light-sensitive tissue that lines the back of the eye
(retina ). Affected individuals at risk often have developmental delays, intellectual disabilities, seizures, or other neurological problems. In the absence of stroke or another brain abnormality, most people with incontinentia pigmenti have normal intelligence.
). Affected individuals at risk often have developmental delays, intellectual disabilities, seizures, or other neurological problems. In the absence of stroke or another brain abnormality, most people with incontinentia pigmenti have normal intelligence.
Other signs and symptoms of incontinentia pigmenti can include hair loss (alopecia) on the scalp and other parts of the body, dental abnormalities (such as small teeth or few teeth), and lined
or few teeth), and lined or pitted
or pitted fingernails and toenails. The features of incontinentia pigmenti may be mild or gone by the time affected individuals reach adulthood.
fingernails and toenails. The features of incontinentia pigmenti may be mild or gone by the time affected individuals reach adulthood.
Frequency
Incontinentia pigmenti is estimated to affect 1.2 in 100,000 individuals worldwide. Between 900 and 1,200 affected individuals have been reported in the scientific literature. Most of the individuals affected are female, but several dozen males with incontinentia pigmenti have also been identified.
Causes
Variants (also called mutations) in the IKBKG gene cause incontinentia pigmenti. The IKBKG gene provides instructions for making a protein that helps regulate nuclear factor-kappa-B. Nuclear factor-kappa-B is a group of related proteins that helps protect cells from self-destructing (undergoing apoptosis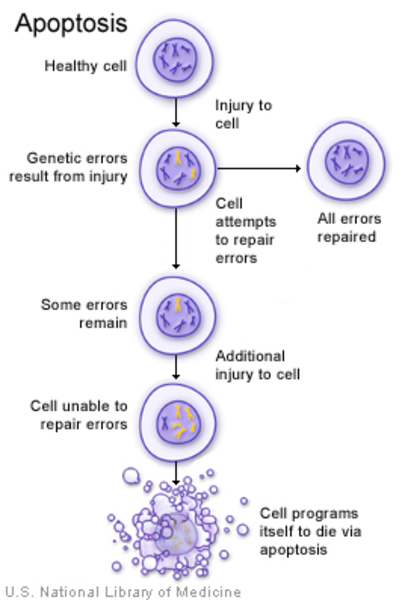 ) in response to certain signals.
) in response to certain signals.
Sixty to 80 percent of affected individuals have a change in the IKBKG gene that deletes some genetic material from the gene. This deletion probably leads to the production of an abnormally small, nonfunctional version of the IKBKG protein. Other people with incontinentia pigmenti have variants that prevent the production of any IKBKG protein. Without this protein, nuclear factor-kappa-B is not regulated properly, and cells are more sensitive to signals that trigger them to self-destruct. Researchers believe that this abnormal cell death leads to the signs and symptoms of incontinentia pigmenti.
Inheritance
This condition is inherited in an X-linked dominant pattern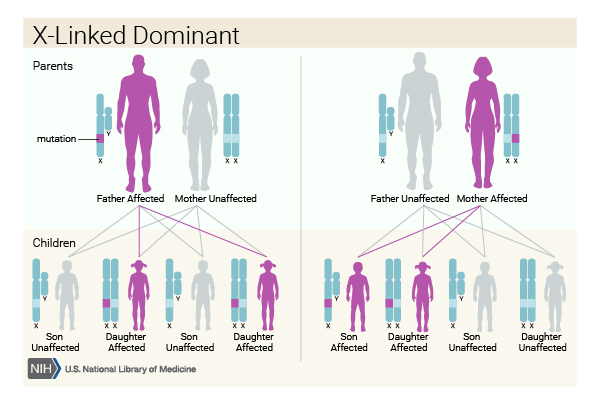 . The gene associated with this condition is located on the X chromosome, which is one of the two sex chromosomes
. The gene associated with this condition is located on the X chromosome, which is one of the two sex chromosomes . In females (who have two X chromosomes), a variant in one of the two copies of the gene in each cell is sufficient to cause the disorder. Some cells produce a normal amount of IKBKG protein, and other cells produce none. The resulting imbalance in cells producing this protein leads to the signs and symptoms of incontinentia pigmenti.
. In females (who have two X chromosomes), a variant in one of the two copies of the gene in each cell is sufficient to cause the disorder. Some cells produce a normal amount of IKBKG protein, and other cells produce none. The resulting imbalance in cells producing this protein leads to the signs and symptoms of incontinentia pigmenti.
In males (who have only one X chromosome), most IKBKG variants result in a total loss of the IKBKG protein. A lack of this protein appears to be lethal early in development, so few males are born with incontinentia pigmenti. Affected males who survive may have an IKBKG variant with relatively mild effects, an IKBKG variant in only some of the body's cells (mosaicism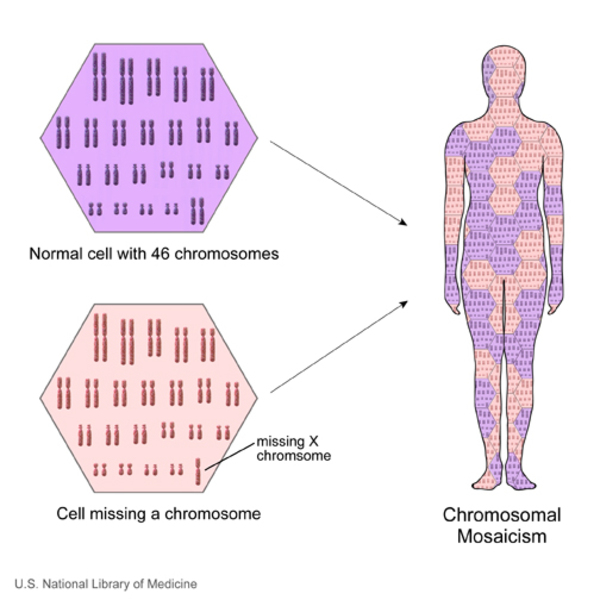 ), or an extra copy of the X chromosome in each cell.
), or an extra copy of the X chromosome in each cell.
Some people with incontinentia pigmenti inherit an IKBKG variant from one affected parent. Other cases result from new variants in the gene and occur in people with no history of the disorder in their family.
Other Names for This Condition
- Bloch-Siemens syndrome
- Bloch-Siemens-Sulzberger Syndrome
- Bloch-Sulzberger Syndrome
- IP
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Ardelean D, Pope E. Incontinentia pigmenti in boys: a series and review of the literature. Pediatr Dermatol. 2006 Nov-Dec;23(6):523-7. doi: 10.1111/j.1525-1470.2006.00302.x. Citation on PubMed
- Berlin AL, Paller AS, Chan LS. Incontinentia pigmenti: a review and update on the molecular basis of pathophysiology. J Am Acad Dermatol. 2002 Aug;47(2):169-87; quiz 188-90. doi: 10.1067/mjd.2002.125949. Citation on PubMed
- Bruckner AL. Incontinentia pigmenti: a window to the role of NF-kappaB function. Semin Cutan Med Surg. 2004 Jun;23(2):116-24. doi: 10.1016/j.sder.2004.01.005. Citation on PubMed
- Goldberg MF. The skin is not the predominant problem in incontinentia pigmenti. Arch Dermatol. 2004 Jun;140(6):748-50. doi: 10.1001/archderm.140.6.748. No abstract available. Citation on PubMed
- Hadj-Rabia S, Froidevaux D, Bodak N, Hamel-Teillac D, Smahi A, Touil Y, Fraitag S, de Prost Y, Bodemer C. Clinical study of 40 cases of incontinentia pigmenti. Arch Dermatol. 2003 Sep;139(9):1163-70. doi: 10.1001/archderm.139.9.1163. Citation on PubMed
- Happle R. A fresh look at incontinentia pigmenti. Arch Dermatol. 2003 Sep;139(9):1206-8. doi: 10.1001/archderm.139.9.1206. No abstract available. Citation on PubMed
- Nelson DL. NEMO, NFkappaB signaling and incontinentia pigmenti. Curr Opin Genet Dev. 2006 Jun;16(3):282-8. doi: 10.1016/j.gde.2006.04.013. Epub 2006 May 2. Citation on PubMed
- Scheuerle AE, Ursini MV. Incontinentia Pigmenti. 1999 Jun 8 [updated 2025 Apr 3]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1472/ Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.