

Description
Hypophosphatasia is an inherited disorder that affects the development of bones and teeth. This condition disrupts a process called mineralization, in which minerals such as calcium and phosphorus are deposited in developing bones and teeth. Mineralization is critical for the formation of bones that are strong and rigid and teeth that can withstand chewing and grinding.
The signs and symptoms of hypophosphatasia vary widely and can appear anywhere from before birth to adulthood. The most severe forms of the disorder tend to occur before birth and in early infancy. Hypophosphatasia weakens and softens the bones, causing skeletal abnormalities similar to another childhood bone disorder called rickets. Affected infants are born with short limbs, an abnormally shaped chest, and soft skull bones. Additional complications in infancy include poor feeding and a failure to gain weight, respiratory problems, and high levels of calcium in the blood (hypercalcemia), which can lead to recurrent vomiting and kidney problems. These complications are life-threatening in some cases.
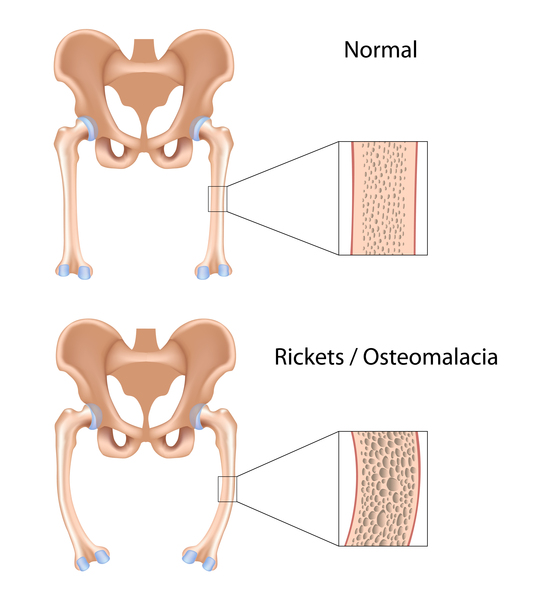
The forms of hypophosphatasia that appear in childhood or adulthood are typically less severe than those that appear in infancy. Early loss of primary (baby) teeth is one of the first signs of the condition in children. Affected children may have short stature with bowed legs or knock knees, enlarged wrist and ankle joints, and an abnormal skull shape. Adult forms of hypophosphatasia are characterized by a softening of the bones known as osteomalacia. In adults, recurrent fractures in the foot and thigh bones can lead to chronic pain. Affected adults may lose their secondary (adult) teeth prematurely and are at increased risk for joint pain and inflammation.
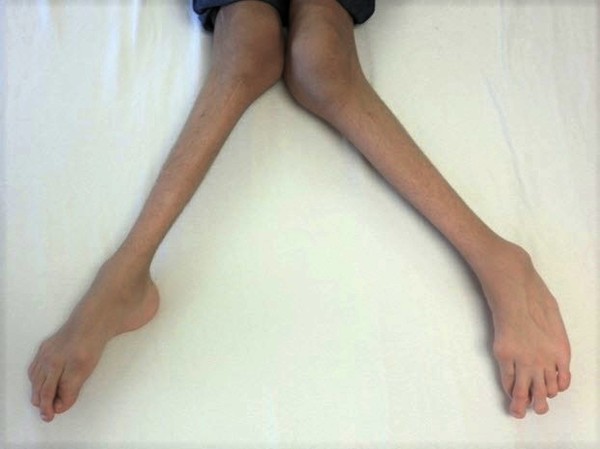
The mildest form of this condition, called odontohypophosphatasia, only affects the teeth. People with this disorder typically experience abnormal tooth development and premature tooth loss, but do not have the skeletal abnormalities seen in other forms of hypophosphatasia.
Frequency
Severe forms of hypophosphatasia affect an estimated 1 in 100,000 newborns. Milder cases, such as those that appear in childhood or adulthood, probably occur more frequently.
Hypophosphatasia has been reported worldwide in people of various ethnic backgrounds. This condition appears to be most common in white populations. It is particularly frequent in a Mennonite population in Manitoba, Canada, where about 1 in 2,500 infants is born with severe features of the condition.
Causes
Mutations in the ALPL gene cause hypophosphatasia. This gene provides instructions for making an enzyme called tissue-nonspecific alkaline phosphatase (TNSALP), which plays an essential role in mineralization of the skeleton and teeth. Mutations in the ALPL gene lead to the production of an abnormal version of TNSALP that cannot participate effectively in the mineralization process. A shortage of TNSALP allows several other substances, which are normally processed by the enzyme, to build up abnormally in the body. Researchers believe that a buildup of one of these compounds, inorganic pyrophosphate (PPi), underlies the defective mineralization of bones and teeth in people with hypophosphatasia.
ALPL gene mutations that almost completely eliminate the activity of TNSALP usually result in the more severe forms of hypophosphatasia. Other mutations, which reduce but do not eliminate the activity of the enzyme, often cause the milder forms of the condition.
Inheritance
The severe forms of hypophosphatasia that appear early in life are inherited in an autosomal recessive pattern. Autosomal recessive inheritance means that two copies of the gene in each cell are altered. Most often, the parents of an individual with an autosomal recessive disorder each carry one copy of the altered gene but do not show signs and symptoms of the disorder.

Milder forms of hypophosphatasia can have either an autosomal recessive or an autosomal dominant pattern of inheritance. Autosomal dominant inheritance means that one copy of the altered gene in each cell is sufficient to cause the disorder.

Other Names for This Condition
- Deficiency of alkaline phosphatase
- Phosphoethanolaminuria
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Khandwala HM, Mumm S, Whyte MP. Low serum alkaline phosphatase activity and pathologic fracture: case report and brief review of hypophosphatasia diagnosed in adulthood. Endocr Pract. 2006 Nov-Dec;12(6):676-81. doi: 10.4158/EP.12.6.676. Citation on PubMed
- Mornet E. Hypophosphatasia: the mutations in the tissue-nonspecific alkaline phosphatase gene. Hum Mutat. 2000;15(4):309-15. doi: 10.1002/(SICI)1098-1004(200004)15:43.0.CO;2-C. Citation on PubMed
- Rockman-Greenberg C. Hypophosphatasia. Pediatr Endocrinol Rev. 2013 Jun;10 Suppl 2:380-8. Citation on PubMed
- Whyte MP. Hypophosphatasia - aetiology, nosology, pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2016 Apr;12(4):233-46. doi: 10.1038/nrendo.2016.14. Epub 2016 Feb 19. Citation on PubMed
- Whyte MP. Hypophosphatasia and the role of alkaline phosphatase in skeletal mineralization. Endocr Rev. 1994 Aug;15(4):439-61. doi: 10.1210/edrv-15-4-439. No abstract available. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.