

Description
Huntington's disease is a progressive brain disorder that causes uncontrolled movements, emotional problems, and loss of thinking ability (cognition).

Adult-onset Huntington's disease, the most common form of this disorder, usually appears in a person's thirties or forties. Early signs and symptoms can include irritability, depression, small involuntary movements, poor coordination, and trouble learning new information or making decisions. Many people with Huntington's disease develop involuntary jerking or twitching movements known as chorea. As the disease progresses, these movements become more pronounced. Affected individuals may have trouble walking, speaking, and swallowing. People with this disorder also experience changes in personality and a decline in thinking and reasoning abilities. Individuals with the adult-onset form of Huntington's disease usually live about 15 to 20 years after signs and symptoms begin.
A less common form of Huntington's disease known as the juvenile form begins in childhood or adolescence. It also involves movement problems and mental and emotional changes. Additional signs of the juvenile form include slow movements, clumsiness, frequent falling, rigidity, slurred speech, and drooling. School performance declines as thinking and reasoning abilities become impaired. Seizures occur in 30 percent to 50 percent of children with this condition. Juvenile Huntington's disease tends to progress more quickly than the adult-onset form; affected individuals usually live 10 to 15 years after signs and symptoms appear.
Frequency
Huntington's disease affects an estimated 3 to 7 per 100,000 people of European ancestry. The disorder appears to be less common in some other populations, including people of Japanese, Chinese, and African descent.
Causes
Variants (also called mutations) in the HTT gene cause Huntington's disease. The HTT gene provides instructions for making a protein called huntingtin. Although the function of this protein is unclear, it appears to play an important role in nerve cells (neurons) in the brain.
The HTT variant that causes Huntington's disease involves a DNA segment known as a CAG trinucleotide repeat. This segment is made up of a series of three DNA building blocks (cytosine, adenine, and guanine) that appear multiple times in a row. Normally, the CAG segment is repeated 10 to 35 times within the gene. In people with Huntington's disease, the CAG segment is repeated 36 to more than 120 times. People with 36 to 39 CAG repeats may or may not develop the signs and symptoms of Huntington's disease, while people with 40 or more repeats almost always develop the disorder.
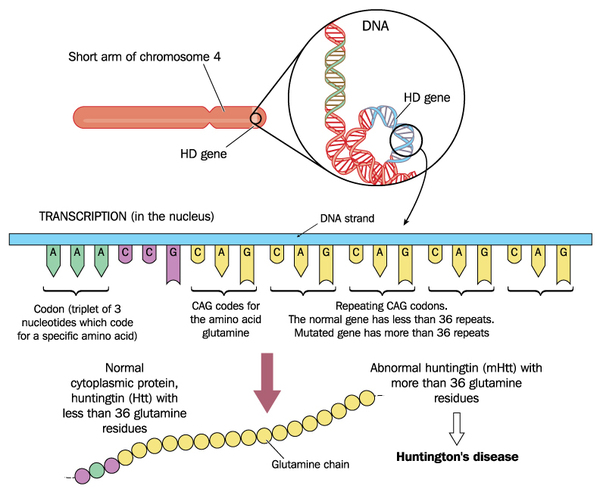
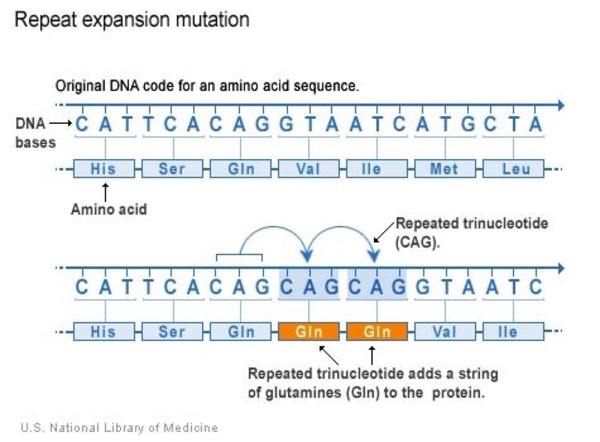
An increase in the size of the CAG segment leads to the production of an abnormally long version of the huntingtin protein. The elongated protein is cut into smaller, toxic fragments that bind together and accumulate in neurons, disrupting the normal functions of these cells. The dysfunction and eventual death of neurons in certain areas of the brain underlie the signs and symptoms of Huntington's disease.
Inheritance
This condition is inherited in an autosomal dominant pattern , which means one copy of the altered gene in each cell is sufficient to cause the disorder. An affected person usually inherits the altered gene from one affected parent. In rare cases, an individual with Huntington's disease does not have a parent with the disorder.
, which means one copy of the altered gene in each cell is sufficient to cause the disorder. An affected person usually inherits the altered gene from one affected parent. In rare cases, an individual with Huntington's disease does not have a parent with the disorder.
As the altered HTT gene is passed from one generation to the next, the size of the CAG trinucleotide repeat often increases in size. A larger number of repeats is usually associated with an earlier onset of signs and symptoms. This phenomenon is called anticipation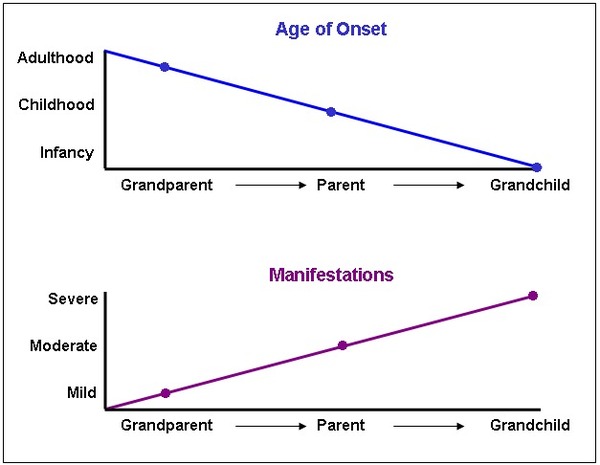 . People with the adult-onset form of Huntington's disease typically have 40 to 50 CAG repeats in the HTT gene, while people with the juvenile form of the disorder tend to have more than 60 CAG repeats.
. People with the adult-onset form of Huntington's disease typically have 40 to 50 CAG repeats in the HTT gene, while people with the juvenile form of the disorder tend to have more than 60 CAG repeats.
Individuals who have 27 to 35 CAG repeats in the HTT gene do not develop Huntington's disease, but they are at risk of having children who will develop the disorder. As the gene is passed from parent to child, the size of the CAG trinucleotide repeat may lengthen into the range associated with Huntington's disease (36 repeats or more).
Other Names for This Condition
- Huntington chorea
- Huntington chronic progressive hereditary chorea
- Huntington disease
- Huntington's chorea
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Bates GP. History of genetic disease: the molecular genetics of Huntington disease - a history. Nat Rev Genet. 2005 Oct;6(10):766-73. doi: 10.1038/nrg1686. Citation on PubMed
- Caldeira Bras I, Dawson J, Kay C, Caron NS, Hayden MR. Huntington Disease. 1998 Oct 23 [updated 2026 Feb 12]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1305/ Citation on PubMed
- Gonzalez-Alegre P, Afifi AK. Clinical characteristics of childhood-onset (juvenile) Huntington disease: report of 12 patients and review of the literature. J Child Neurol. 2006 Mar;21(3):223-9. doi: 10.2310/7010.2006.00055. Citation on PubMed
- Imarisio S, Carmichael J, Korolchuk V, Chen CW, Saiki S, Rose C, Krishna G, Davies JE, Ttofi E, Underwood BR, Rubinsztein DC. Huntington's disease: from pathology and genetics to potential therapies. Biochem J. 2008 Jun 1;412(2):191-209. doi: 10.1042/BJ20071619. Citation on PubMed
- Jones L, Hughes A. Pathogenic mechanisms in Huntington's disease. Int Rev Neurobiol. 2011;98:373-418. doi: 10.1016/B978-0-12-381328-2.00015-8. Citation on PubMed
- Kent A. Huntington's disease. Nurs Stand. 2004 Apr 21-27;18(32):45-51; quiz 52-3. doi: 10.7748/ns2004.04.18.32.45.c3596. Citation on PubMed
- Maiuri T, Mocle AJ, Hung CL, Xia J, van Roon-Mom WM, Truant R. Huntingtin is a scaffolding protein in the ATM oxidative DNA damage response complex. Hum Mol Genet. 2017 Jan 15;26(2):395-406. doi: 10.1093/hmg/ddw395. Citation on PubMed
- Sturrock A, Leavitt BR. The clinical and genetic features of Huntington disease. J Geriatr Psychiatry Neurol. 2010 Dec;23(4):243-59. doi: 10.1177/0891988710383573. Epub 2010 Oct 5. Citation on PubMed
- Tost H, Wendt CS, Schmitt A, Heinz A, Braus DF. Huntington's disease: phenomenological diversity of a neuropsychiatric condition that challenges traditional concepts in neurology and psychiatry. Am J Psychiatry. 2004 Jan;161(1):28-34. doi: 10.1176/appi.ajp.161.1.28. No abstract available. Citation on PubMed
- Walker FO. Huntington's disease. Lancet. 2007 Jan 20;369(9557):218-28. doi: 10.1016/S0140-6736(07)60111-1. Citation on PubMed
- Young AB. Huntingtin in health and disease. J Clin Invest. 2003 Feb;111(3):299-302. doi: 10.1172/JCI17742. No abstract available. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.