

Description
Histiocytosis-lymphadenopathy plus syndrome (also known as SLC29A3 spectrum disorder) is a group of conditions with overlapping signs and symptoms that affect many parts of the body. This group of disorders includes H syndrome, pigmented hypertrichosis with insulin-dependent diabetes mellitus (PHID), Faisalabad histiocytosis, and familial Rosai-Dorfman disease (RDD). These conditions were once thought to be distinct disorders; however, because of the overlapping features and shared genetic cause, they are now considered to be part of the same disease spectrum. While some affected individuals have signs and symptoms characteristic of one of the conditions, others have a range of features from two or more of the conditions. The pattern of signs and symptoms can vary even within the same family.
A feature common to the disorders in this spectrum is histiocytosis, which is the overgrowth of immune system cells called histiocytes. The cells abnormally accumulate in one or more tissues in the body, which can lead to organ or tissue damage. The buildup often occurs in the lymph nodes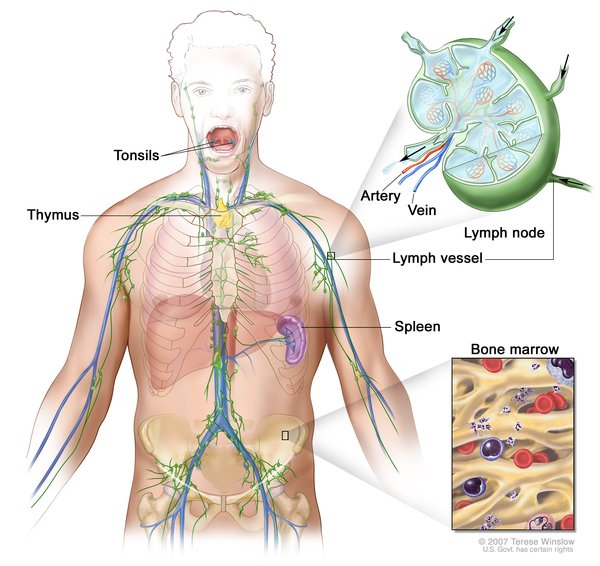 , leading to swelling of the lymph nodes (lymphadenopathy). Other areas of cell accumulation can include the skin
, leading to swelling of the lymph nodes (lymphadenopathy). Other areas of cell accumulation can include the skin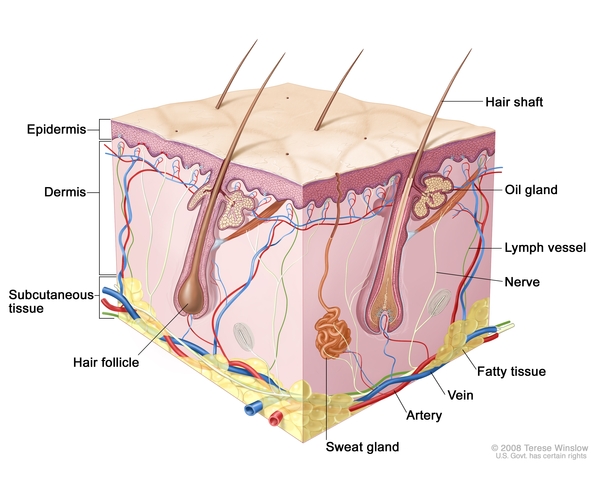 , kidneys
, kidneys , brain and spinal cord (central nervous system
, brain and spinal cord (central nervous system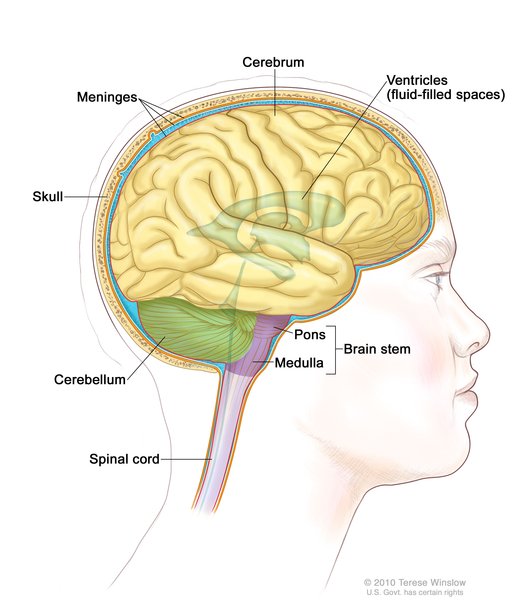 ), or digestive tract
), or digestive tract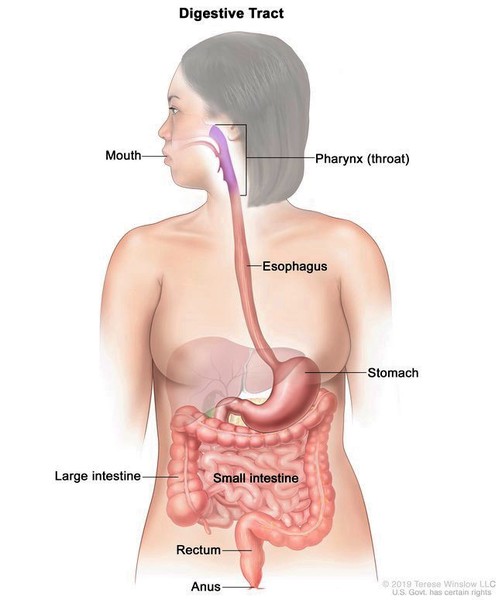 .
.
This spectrum is known as histiocytosis-lymphadenopathy plus syndrome because the disorders that make up the spectrum can have additional signs and symptoms. A characteristic feature of H syndrome is abnormal patches of skin (lesions), typically on the lower body. These lesions are unusually dark (hyperpigmented) and have excessive hair growth (hypertrichosis). In addition, histiocytes accumulate at the site of the skin lesions. Other features of H syndrome include enlargement of the liver (hepatomegaly), heart abnormalities, hearing loss, reduced amounts of hormones that direct sexual development (hypogonadism), and short stature.
Like H syndrome, PHID causes patches of hyperpigmented skin with hypertrichosis. PHID is also characterized by the development of type 1 diabetes (also known as insulin-dependent diabetes mellitus), which usually begins in childhood. Type 1 diabetes occurs when the body does not produce enough of the hormone insulin, leading to dysregulation of levels of blood glucose, also called blood sugar.
Faisalabad histiocytosis typically causes lymphadenopathy and swelling of the eyelids due to accumulation of histiocytes. Affected individuals can also have joint deformities called contractures in their fingers or toes and hearing loss.
The most common feature of familial RDD is lymphadenopathy, usually affecting lymph nodes in the neck. Histiocytes can also accumulate in other parts of the body. (Familial RDD is one of several forms of RDD; the other forms are not considered part of histiocytosis-lymphadenopathy plus syndrome.)
Frequency
Histiocytosis-lymphadenopathy plus syndrome is a rare disorder, affecting approximately 100 individuals worldwide.
Causes
Histiocytosis-lymphadenopathy plus syndrome is caused by variants (also known as mutations) in the SLC29A3 gene, which provides instructions for making a protein called equilibrative nucleoside transporter 3 (ENT3). ENT3 belongs to a family of proteins that transport molecules called nucleosides in cells. With chemical modification, nucleosides become the building blocks of DNA, its chemical cousin RNA, and molecules such as ATP and GTP, which serve as energy sources in the cell. Molecules derived from nucleosides play an important role in many functions throughout the body.
ENT3 is found in cellular structures called lysosomes , which break down large molecules into smaller ones that can be reused by cells. Researchers believe that this protein transports nucleosides generated by the breakdown of DNA and RNA out of lysosomes into the cell so they can be reused. The protein is also thought to transport nucleosides into structures called mitochondria
, which break down large molecules into smaller ones that can be reused by cells. Researchers believe that this protein transports nucleosides generated by the breakdown of DNA and RNA out of lysosomes into the cell so they can be reused. The protein is also thought to transport nucleosides into structures called mitochondria , which are the energy-producing centers of cells. In mitochondria, nucleosides are likely used in the formation or repair of DNA found in these structures, known as mitochondrial DNA.
, which are the energy-producing centers of cells. In mitochondria, nucleosides are likely used in the formation or repair of DNA found in these structures, known as mitochondrial DNA.
The SLC29A3 gene variants involved in histiocytosis-lymphadenopathy plus syndrome reduce or eliminate the activity of the ENT3 protein. Researchers speculate that the resulting impairment of nucleoside transport leads to a buildup of nucleosides in lysosomes, which may be damaging to cell function. A lack of ENT3 activity may also lead to a reduction in the amount of nucleosides in mitochondria. This nucleoside shortage could impair cellular energy production, which would impact many body systems. It is unclear how the variants lead to histiocytosis and other features of the condition or why affected individuals can have different patterns of signs and symptoms.
Inheritance
This condition is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell have variants. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
, which means both copies of the gene in each cell have variants. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
Other Names for This Condition
- SLC29A3 disorder
- SLC29A3 spectrum disorder
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Abla O, Jacobsen E, Picarsic J, Krenova Z, Jaffe R, Emile JF, Durham BH, Braier J, Charlotte F, Donadieu J, Cohen-Aubart F, Rodriguez-Galindo C, Allen C, Whitlock JA, Weitzman S, McClain KL, Haroche J, Diamond EL. Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-Destombes disease. Blood. 2018 Jun 28;131(26):2877-2890. doi: 10.1182/blood-2018-03-839753. Epub 2018 May 2. Citation on PubMed
- Bolze A, Abhyankar A, Grant AV, Patel B, Yadav R, Byun M, Caillez D, Emile JF, Pastor-Anglada M, Abel L, Puel A, Govindarajan R, de Pontual L, Casanova JL. A mild form of SLC29A3 disorder: a frameshift deletion leads to the paradoxical translation of an otherwise noncoding mRNA splice variant. PLoS One. 2012;7(1):e29708. doi: 10.1371/journal.pone.0029708. Epub 2012 Jan 4. Citation on PubMed or Free article on PubMed Central
- Kang N, Jun AH, Bhutia YD, Kannan N, Unadkat JD, Govindarajan R. Human equilibrative nucleoside transporter-3 (hENT3) spectrum disorder mutations impair nucleoside transport, protein localization, and stability. J Biol Chem. 2010 Sep 3;285(36):28343-52. doi: 10.1074/jbc.M110.109199. Epub 2010 Jul 1. Citation on PubMed or Free article on PubMed Central
- Morgan NV, Morris MR, Cangul H, Gleeson D, Straatman-Iwanowska A, Davies N, Keenan S, Pasha S, Rahman F, Gentle D, Vreeswijk MP, Devilee P, Knowles MA, Ceylaner S, Trembath RC, Dalence C, Kismet E, Koseoglu V, Rossbach HC, Gissen P, Tannahill D, Maher ER. Mutations in SLC29A3, encoding an equilibrative nucleoside transporter ENT3, cause a familial histiocytosis syndrome (Faisalabad histiocytosis) and familial Rosai-Dorfman disease. PLoS Genet. 2010 Feb 5;6(2):e1000833. doi: 10.1371/journal.pgen.1000833. Citation on PubMed or Free article on PubMed Central
- Spiegel R, Cliffe ST, Buckley MF, Crow YJ, Urquhart J, Horovitz Y, Tenenbaum-Rakover Y, Newman WG, Donnai D, Shalev SA. Expanding the clinical spectrum of SLC29A3 gene defects. Eur J Med Genet. 2010 Sep-Oct;53(5):309-13. doi: 10.1016/j.ejmg.2010.06.012. Epub 2010 Jul 7. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.