

Description
Hereditary sensory and autonomic neuropathy type II (HSAN2) is a condition that primarily affects the sensory nerve cells (sensory neurons), which transmit information about sensations such as pain, temperature, and touch to the brain. These sensations are impaired in people with HSAN2. In some affected people, the condition may also cause mild abnormalities of the autonomic neurons, which control involuntary body functions such as heart rate, digestion, and breathing. The sensory and autonomic neurons are part of the body's peripheral nervous system, which comprises the nerves outside the brain and spinal cord. HSAN2 is considered a form of peripheral neuropathy.
The signs and symptoms of HSAN2 typically begin in infancy or early childhood. The first sign of the condition is usually numbness in the hands and feet. Soon after, affected individuals lose the ability to feel pain or sense hot and cold. In people with HSAN2, unnoticed injuries often lead to open sores (ulcers) on the hands and feet. Because affected individuals cannot feel the pain of these sores, they may not seek treatment right away. Without treatment, the ulcers can become infected and may require amputation of the affected area. People with HSAN2 often injure themselves unintentionally, typically by biting the tongue, lips, or fingers. These injuries may lead to loss of the affected areas, such as the tip of the tongue. Affected individuals often have injuries and fractures in their hands, feet, limbs, and joints that go untreated because of the inability to feel pain. Repeated injury can lead to a condition called Charcot joints, in which the bones and tissue surrounding joints are damaged.
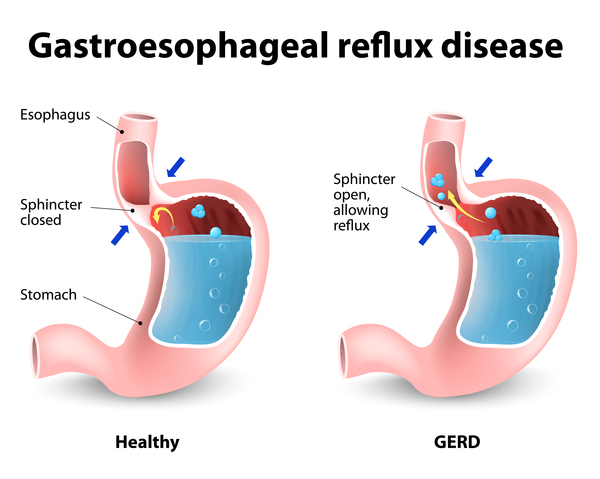
The effects of HSAN2 on the autonomic nervous system are more variable. Some infants with HSAN2 have digestive problems such as the backflow of stomach acids into the esophagus (gastroesophageal reflux) or slow eye-blink or gag reflexes. Affected individuals may also have weak deep-tendon reflexes, such as the reflex being tested when a doctor taps the knee with a hammer.
Some people with HSAN2 lose a type of taste bud on the tip of the tongue called lingual fungiform papillae and have a diminished sense of taste.
Frequency
HSAN2 is a rare disease; however, the prevalence is unknown.
Causes
There are several types of HSAN2, each caused by mutations in a different gene. HSAN2A is caused by mutations in the WNK1 gene, and HSAN2B is caused by mutations in the RETREG1 gene. Additional types caused by mutations in other genes are rare. Although different genes are involved, all types of HSAN2 have similar signs and symptoms.
The WNK1 gene provides instructions for making multiple versions (isoforms) of the WNK1 protein. HSAN2A is caused by mutations that affect a particular isoform called the WNK1/HSN2 protein. This protein is found in the cells of the nervous system, including sensory neurons. The mutations involved in HSAN2A result in an abnormally short WNK1/HSN2 protein. Although the function of this protein is not well understood, it is likely that the abnormally short version cannot function properly or is broken down. People with HSAN2A have a reduction in the number of sensory neurons; however, the role that WNK1/HSN2 protein changes play in that loss is unclear.
HSAN2B is caused by mutations in the RETREG1 gene. These mutations lead to an abnormally short and nonfunctional protein. The RETREG1 protein is normally found in sensory and autonomic neurons. It is involved in the recycling of worn-out cell parts (autophagy), specifically a cell structure called the endoplasmic reticulum. When the RETREG1 protein is nonfunctional, recycling of the endoplasmic reticulum is impaired. The buildup of these structures likely results in death of the neurons.

The loss of neurons leads to the inability to feel pain, temperature, and touch sensations and to the impairment of the autonomic nervous system seen in people with HSAN2.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Congenital sensory neuropathy
- Hereditary sensory and autonomic neuropathy type 2
- HSAN type II
- HSAN2
- HSAN2A
- HSAN2B
- HSAN2C
- HSAN2D
- HSANII
- HSN type II
- Morvan disease
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Axelrod FB, Gold-von Simson G. Hereditary sensory and autonomic neuropathies: types II, III, and IV. Orphanet J Rare Dis. 2007 Oct 3;2:39. doi: 10.1186/1750-1172-2-39. Citation on PubMed or Free article on PubMed Central
- Davidson G, Murphy S, Polke J, Laura M, Salih M, Muntoni F, Blake J, Brandner S, Davies N, Horvath R, Price S, Donaghy M, Roberts M, Foulds N, Ramdharry G, Soler D, Lunn M, Manji H, Davis M, Houlden H, Reilly M. Frequency of mutations in the genes associated with hereditary sensory and autonomic neuropathy in a UK cohort. J Neurol. 2012 Aug;259(8):1673-85. doi: 10.1007/s00415-011-6397-y. Citation on PubMed or Free article on PubMed Central
- Huang CL, Kuo E. Mechanisms of disease: WNK-ing at the mechanism of salt-sensitive hypertension. Nat Clin Pract Nephrol. 2007 Nov;3(11):623-30. doi: 10.1038/ncpneph0638. Citation on PubMed
- Khaminets A, Heinrich T, Mari M, Grumati P, Huebner AK, Akutsu M, Liebmann L, Stolz A, Nietzsche S, Koch N, Mauthe M, Katona I, Qualmann B, Weis J, Reggiori F, Kurth I, Hubner CA, Dikic I. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature. 2015 Jun 18;522(7556):354-8. doi: 10.1038/nature14498. Epub 2015 Jun 3. Citation on PubMed
- Kurth I, Pamminger T, Hennings JC, Soehendra D, Huebner AK, Rotthier A, Baets J, Senderek J, Topaloglu H, Farrell SA, Nurnberg G, Nurnberg P, De Jonghe P, Gal A, Kaether C, Timmerman V, Hubner CA. Mutations in FAM134B, encoding a newly identified Golgi protein, cause severe sensory and autonomic neuropathy. Nat Genet. 2009 Nov;41(11):1179-81. doi: 10.1038/ng.464. Epub 2009 Oct 18. Citation on PubMed
- Lafreniere RG, MacDonald ML, Dube MP, MacFarlane J, O'Driscoll M, Brais B, Meilleur S, Brinkman RR, Dadivas O, Pape T, Platon C, Radomski C, Risler J, Thompson J, Guerra-Escobio AM, Davar G, Breakefield XO, Pimstone SN, Green R, Pryse-Phillips W, Goldberg YP, Younghusband HB, Hayden MR, Sherrington R, Rouleau GA, Samuels ME. Identification of a novel gene (HSN2) causing hereditary sensory and autonomic neuropathy type II through the Study of Canadian Genetic Isolates. Am J Hum Genet. 2004 May;74(5):1064-73. doi: 10.1086/420795. Epub 2004 Apr 1. Citation on PubMed or Free article on PubMed Central
- Murphy SM, Davidson GL, Brandner S, Houlden H, Reilly MM. Mutation in FAM134B causing severe hereditary sensory neuropathy. J Neurol Neurosurg Psychiatry. 2012 Jan;83(1):119-20. doi: 10.1136/jnnp.2010.228965. Epub 2010 Nov 28. Citation on PubMed or Free article on PubMed Central
- Riviere JB, Ramalingam S, Lavastre V, Shekarabi M, Holbert S, Lafontaine J, Srour M, Merner N, Rochefort D, Hince P, Gaudet R, Mes-Masson AM, Baets J, Houlden H, Brais B, Nicholson GA, Van Esch H, Nafissi S, De Jonghe P, Reilly MM, Timmerman V, Dion PA, Rouleau GA. KIF1A, an axonal transporter of synaptic vesicles, is mutated in hereditary sensory and autonomic neuropathy type 2. Am J Hum Genet. 2011 Aug 12;89(2):219-30. doi: 10.1016/j.ajhg.2011.06.013. Epub 2011 Aug 4. Citation on PubMed or Free article on PubMed Central
- Shekarabi M, Girard N, Riviere JB, Dion P, Houle M, Toulouse A, Lafreniere RG, Vercauteren F, Hince P, Laganiere J, Rochefort D, Faivre L, Samuels M, Rouleau GA. Mutations in the nervous system--specific HSN2 exon of WNK1 cause hereditary sensory neuropathy type II. J Clin Invest. 2008 Jul;118(7):2496-505. doi: 10.1172/JCI34088. Citation on PubMed or Free article on PubMed Central
- Verpoorten N, De Jonghe P, Timmerman V. Disease mechanisms in hereditary sensory and autonomic neuropathies. Neurobiol Dis. 2006 Feb;21(2):247-55. doi: 10.1016/j.nbd.2005.08.004. Epub 2005 Sep 23. Citation on PubMed
- Yuan J, Matsuura E, Higuchi Y, Hashiguchi A, Nakamura T, Nozuma S, Sakiyama Y, Yoshimura A, Izumo S, Takashima H. Hereditary sensory and autonomic neuropathy type IID caused by an SCN9A mutation. Neurology. 2013 Apr 30;80(18):1641-9. doi: 10.1212/WNL.0b013e3182904fdd. Epub 2013 Apr 17. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.