

Description
X-linked hypophosphatemia is characterized by low levels of phosphate in the blood (hypophosphatemia), which can lead to skeletal abnormalities. Phosphate is a mineral that is essential for the normal development of bones and teeth. Without adequate amounts of phosphate, the bones can weaken or soften, leading to a condition called rickets in growing children or a similar condition called osteomalacia in adults.
The features of X-linked hypophosphatemia vary widely, even among members of the same family. Mildly affected individuals may have hypophosphatemia without other signs and symptoms. More severely affected individuals typically experience slow growth beginning in early childhood, and they may be shorter than their peers. They often develop bone abnormalities that can impair movement and cause pain, such as legs that are abnormally curved (bowed) because the bones are too weak to bear weight. These abnormalities tend to worsen over time.
In adults with X-linked hypophosphatemia, osteomalacia can make the bones more prone to a type of fracture called a stress fracture, which can be painful. Affected adults may also have abnormal calcium deposits that occur near the joints where the ligaments and tendons attach (enthesopathy) and a painful joint disorder called osteoarthritis.
Other signs and symptoms of X-linked hypophosphatemia can include premature fusion of the skull bones (craniosynostosis) and hearing loss. Affected individuals may also have dental abnormalities, which can include pus caused by a bacterial infection (abscess). In rare cases, people with X-linked hypophosphatemia have abnormalities of the spine, which can include a narrowing of the spinal canal that can pinch the upper part of the spinal cord (spinal stenosis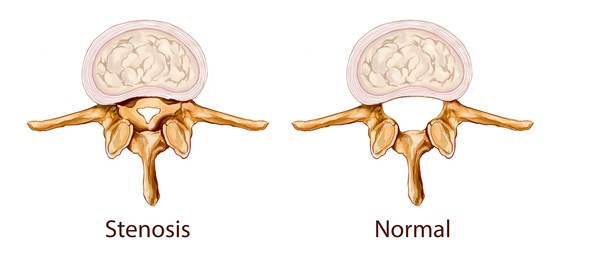 ) and the formation of fluid-filled cysts (syrinx) within the spinal cord.
) and the formation of fluid-filled cysts (syrinx) within the spinal cord.
With early treatment, people with X-linked hypophosphatemia may have less-severe skeletal signs and symptoms and fewer long-term health issues.
Frequency
X-linked hypophosphatemia affects 1 in 20,000 to 25,000 newborns.
Causes
Variants (also called mutations) in the PHEX gene cause X-linked hypophoshatemia. The PHEX gene provides instructions for making an enzyme that appears to be important for the proper development of bones and teeth.
The PHEX gene variants that cause X-linked hypophosphatemia lead to an increase in the amount of fibroblast growth factor 23. Fibroblast growth factor 23 is a protein that works within the intestines and kidneys to keep the body’s phosphate levels in balance. The intestines normally absorb phosphate from food and release it into the bloodstream, while the kidneys release excess phosphate in the urine. When more phosphate is needed, the kidneys can also reabsorb phosphate into the bloodstream. When PHEX gene variants increase the amount of fibroblast growth factor 23, the absorption of phosphate in the intestines and the reabsorption of phosphate by the kidneys are impaired. As a result, less phosphate is available for normal bone development and maintenance, which contributes to the skeletal abnormalities seen in people with X-linked hypophosphatemia.
Inheritance
This condition is inherited in an X-linked pattern. A condition is considered X-linked if the altered gene that causes the disorder is located on the X chromosome, one of the two sex chromosomes in each cell. In males (who have only one X chromosome), a variant in the only copy of the gene in each cell is typically sufficient to cause the condition. This is not always the case for X-linked disorders in females (who have two X chromosomes in each cell). However, for X-linked hypophosphatemia, females with one PHEX gene variant typically have signs and symptoms that are similar to those seen in males with one PHEX gene variant.
in each cell. In males (who have only one X chromosome), a variant in the only copy of the gene in each cell is typically sufficient to cause the condition. This is not always the case for X-linked disorders in females (who have two X chromosomes in each cell). However, for X-linked hypophosphatemia, females with one PHEX gene variant typically have signs and symptoms that are similar to those seen in males with one PHEX gene variant.
A characteristic of X-linked conditions is that fathers pass the variant to all of their daughters and none of their sons, while mothers have a 50 percent chance of passing the variant to each child.
Other Names for This Condition
- Hypophosphatemic rickets, PHEX-Related
- X-Linked Hypophosphatemic Rickets
- X-Linked Vitamin D-Resistant Rickets
- XLHR
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Ackah SA, Imel EA. Approach to Hypophosphatemic Rickets. J Clin Endocrinol Metab. 2022 Dec 17;108(1):209-220. doi: 10.1210/clinem/dgac488. Citation on PubMed
- Baroncelli GI, Bertelloni S, Sodini F, Galli L, Vanacore T, Fiore L, Saggese G. Genetic advances, biochemical and clinical features and critical approach to treatment of patients with X-linked hypophosphatemic rickets. Pediatr Endocrinol Rev. 2004 Jun;1(4):361-79. Citation on PubMed
- Bitzan M, Goodyer PR. Hypophosphatemic Rickets. Pediatr Clin North Am. 2019 Feb;66(1):179-207. doi: 10.1016/j.pcl.2018.09.004. Citation on PubMed
- Dahash BA, Sankararaman S. Rickets. 2023 Aug 7. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2026 Jan-. Available from http://www.ncbi.nlm.nih.gov/books/NBK562285/ Citation on PubMed
- Kamenicky P, Briot K, Munns CF, Linglart A. X-linked hypophosphataemia. Lancet. 2024 Aug 31;404(10455):887-901. doi: 10.1016/S0140-6736(24)01305-9. Epub 2024 Aug 21. Citation on PubMed
- Laurent MR, Harvengt P, Mortier GR, Bockenhauer D. X-Linked Hypophosphatemia. 2012 Feb 9 [updated 2023 Dec 14]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK83985/ Citation on PubMed
- Marik B, Bagga A, Sinha A, Khandelwal P, Hari P, Sharma A. Genetic and clinical profile of patients with hypophosphatemic rickets. Eur J Med Genet. 2022 Aug;65(8):104540. doi: 10.1016/j.ejmg.2022.104540. Epub 2022 Jun 21. Citation on PubMed
- Narasimhan S, Lavik A, Auron M. Rickets. Pediatr Rev. 2025 Sep 1;46(9):494-509. doi: 10.1542/pir.2024-006494. Citation on PubMed
- Rothenbuhler A, Schnabel D, Hogler W, Linglart A. Diagnosis, treatment-monitoring and follow-up of children and adolescents with X-linked hypophosphatemia (XLH). Metabolism. 2020 Feb;103S:153892. doi: 10.1016/j.metabol.2019.03.009. Epub 2019 Mar 27. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.