

Description
GM1 gangliosidosis is an inherited disorder that destroys nerve cells (neurons) in the brain and spinal cord. This condition can be classified as one of three major types based on the age at which signs and symptoms first appear. However, the signs and symptoms of these three types can overlap, leading some researchers to believe that GM1 gangliosidosis occurs on a spectrum instead of as three distinct types.
The signs and symptoms of the most severe form of GM1 gangliosidosis, called type I or the infantile form, usually develop by the age of 6 months. Infants with this form of the disorder typically appear normal until their development slows and the muscles used for movement weaken. Affected infants eventually lose the skills they had previously acquired (developmentally regress) and may develop an exaggerated startle reaction to loud noises. Over time, children with GM1 gangliosidosis type I develop an enlarged liver and spleen
and spleen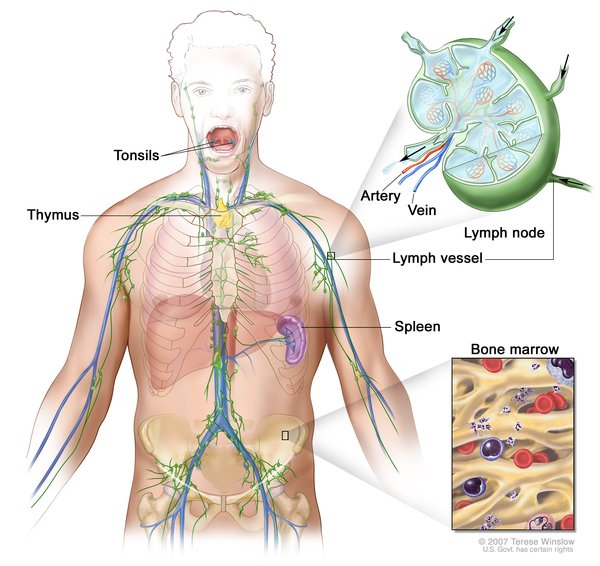 (hepatosplenomegaly) and skeletal abnormalities. Affected children often have seizures and profound intellectual disability.
(hepatosplenomegaly) and skeletal abnormalities. Affected children often have seizures and profound intellectual disability.
People with GM1 gangliosidosis type I can lose their vision due to clouding of the clear outer covering of the eye (the cornea ) and the breakdown of the light-sensing tissue at the back of the eye (the retina
) and the breakdown of the light-sensing tissue at the back of the eye (the retina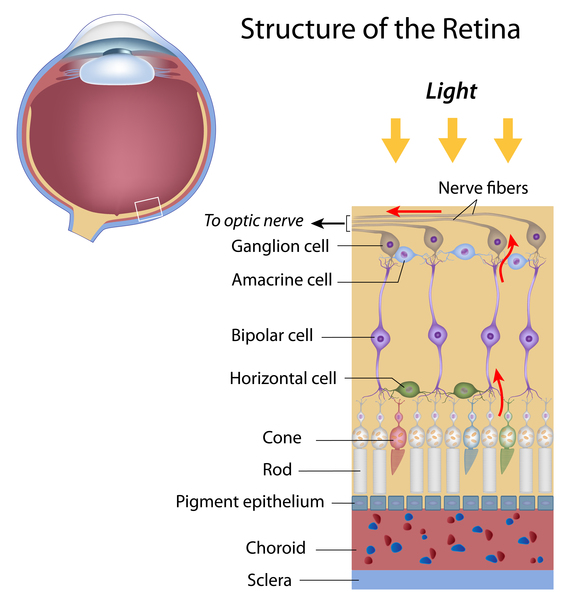 ). Affected individuals also develop a red area in the eye known as a cherry-red spot. In some cases, affected individuals have distinctive facial features that are described as "coarse," enlarged gums (gingival hypertrophy
). Affected individuals also develop a red area in the eye known as a cherry-red spot. In some cases, affected individuals have distinctive facial features that are described as "coarse," enlarged gums (gingival hypertrophy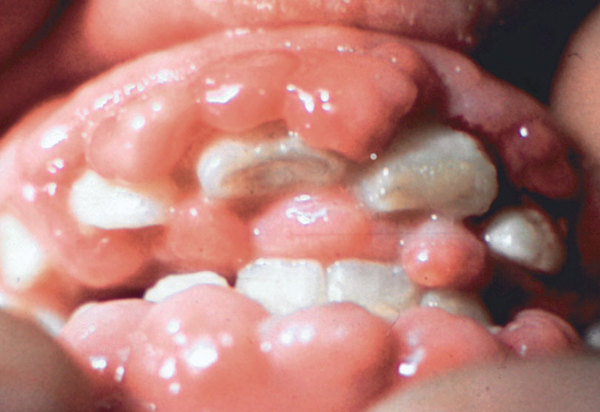 ), and an enlarged and weakened heart muscle (cardiomyopathy). Individuals with type I usually do not survive past early childhood.
), and an enlarged and weakened heart muscle (cardiomyopathy). Individuals with type I usually do not survive past early childhood.
GM1 gangliosidosis type II occurs in one of two forms: the late infantile or the juvenile forms. Children with type II develop normally early in life, but they begin to show signs and symptoms of the condition around the age of 18 months (late infantile form) or 5 years (juvenile form). Individuals with GM1 gangliosidosis type II experience developmental regression but usually do not have cherry-red spots, coarse facial features, or enlarged organs. Type II usually progresses more slowly than type I, but it still shortens life expectancy. People with the late infantile form typically survive into mid-childhood, while those with the juvenile form may live into early adulthood.
GM1 gangliosidosis type III is the adult or chronic form of the condition, and this is the mildest form. The age at which symptoms first appear varies in people with GM1 gangliosidosis type III, although most affected individuals develop signs and symptoms in their teens. The characteristic features of this type include involuntary tensing of various muscles (dystonia) and abnormalities of the spinal bones (vertebrae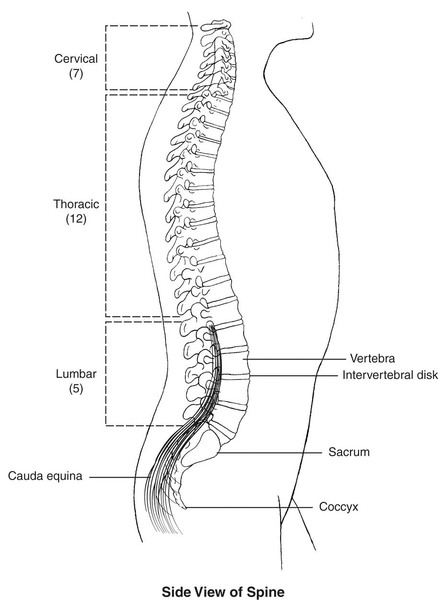 ). Life expectancy varies among people with GM1 gangliosidosis type III.
). Life expectancy varies among people with GM1 gangliosidosis type III.
Frequency
GM1 gangliosidosis is estimated to occur in 1 in 100,000 to 200,000 newborns. Type I is reported more frequently than the other forms of this condition. Most individuals with type III are of Japanese descent.
Causes
Variants (also called mutations) in the GLB1 gene cause GM1 gangliosidosis. The GLB1 gene provides instructions for making an enzyme called beta-galactosidase (β-galactosidase). This enzyme is found in lysosomes , which are compartments within cells that break down and recycle different types of molecules. β-galactosidase helps break down several molecules, including a substance called GM1 ganglioside. GM1 ganglioside is important for normal functioning of neurons in the brain.
, which are compartments within cells that break down and recycle different types of molecules. β-galactosidase helps break down several molecules, including a substance called GM1 ganglioside. GM1 ganglioside is important for normal functioning of neurons in the brain.
The GLB1 gene variants produce versions of β-galactosidase that are not as effective at breaking down GM1 ganglioside as the normal version of the enzyme. Without enough functional β-galactosidase, GM1 ganglioside cannot be broken down when it is no longer needed. This substance builds up to toxic levels in many tissues and organs, particularly in the brain. Damage caused by the buildup of GM1 ganglioside leads to the destruction of neurons, causing many of the signs and symptoms of GM1 gangliosidosis.
In general, people with GM1 gangliosidosis have milder signs and symptoms if they have higher levels of functional β-galactosidase activity. These individuals are still able to break down some amount of GM1 ganglioside, and less of the enzyme builds up inside lysosomes.
Conditions like GM1 gangliosidosis that cause molecules to build up inside lysosomes are called lysosomal storage disorders.
Inheritance
This condition is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
, which means both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
Other Names for This Condition
- Beta-galactosidase-1 (GLB1) deficiency
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Arash-Kaps L, Komlosi K, Seegraber M, Diederich S, Paschke E, Amraoui Y, Beblo S, Dieckmann A, Smitka M, Hennermann JB. The Clinical and Molecular Spectrum of GM1 Gangliosidosis. J Pediatr. 2019 Dec;215:152-157.e3. doi: 10.1016/j.jpeds.2019.08.016. Citation on PubMed
- Bingaman A, Waggoner C, Andrews SM, Pangonis D, Trad M, Giugliani R, Giorgino R, Jarnes J, Vakili R, Ballard V, Peay HL. GM1-gangliosidosis: The caregivers' assessments of symptom impact and most important symptoms to treat. Am J Med Genet A. 2023 Feb;191(2):408-423. doi: 10.1002/ajmg.a.63038. Epub 2022 Dec 21. Citation on PubMed
- Hofer D, Paul K, Fantur K, Beck M, Roubergue A, Vellodi A, Poorthuis BJ, Michelakakis H, Plecko B, Paschke E. Phenotype determining alleles in GM1 gangliosidosis patients bearing novel GLB1 mutations. Clin Genet. 2010 Sep;78(3):236-46. doi: 10.1111/j.1399-0004.2010.01379.x. Epub 2010 Feb 11. Citation on PubMed
- Lang FM, Korner P, Harnett M, Karunakara A, Tifft CJ. The natural history of Type 1 infantile GM1 gangliosidosis: A literature-based meta-analysis. Mol Genet Metab. 2020 Mar;129(3):228-235. doi: 10.1016/j.ymgme.2019.12.012. Epub 2019 Dec 30. Citation on PubMed
- Regier DS, Proia RL, D'Azzo A, Tifft CJ. The GM1 and GM2 Gangliosidoses: Natural History and Progress toward Therapy. Pediatr Endocrinol Rev. 2016 Jun;13 Suppl 1(Suppl 1):663-73. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.