

Description
Glycogen storage disease type 0 (also known as GSD 0) is a condition caused by the body's inability to form a complex sugar called glycogen, which is a major source of stored energy in the body. GSD 0 has two types: in muscle GSD 0, glycogen formation in the muscles is impaired, and in liver GSD 0, glycogen formation in the liver is impaired.

The signs and symptoms of muscle GSD 0 typically begin in early childhood. Affected individuals often experience muscle pain and weakness or episodes of fainting (syncope) following moderate physical activity, such as walking up stairs. The loss of consciousness that occurs with fainting typically lasts up to several hours. Some individuals with muscle GSD 0 have a disruption of the heart's normal rhythm (arrhythmia) known as long QT syndrome. In all affected individuals, muscle GSD 0 impairs the heart's ability to effectively pump blood and increases the risk of cardiac arrest and sudden death, particularly after physical activity. Sudden death from cardiac arrest can occur in childhood or adolescence in people with muscle GSD 0.
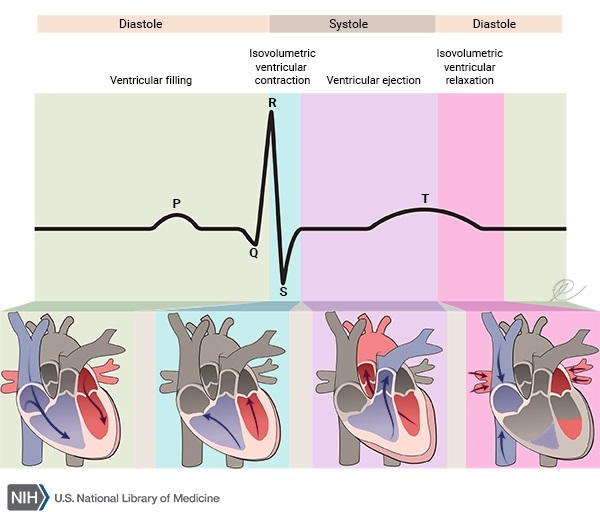
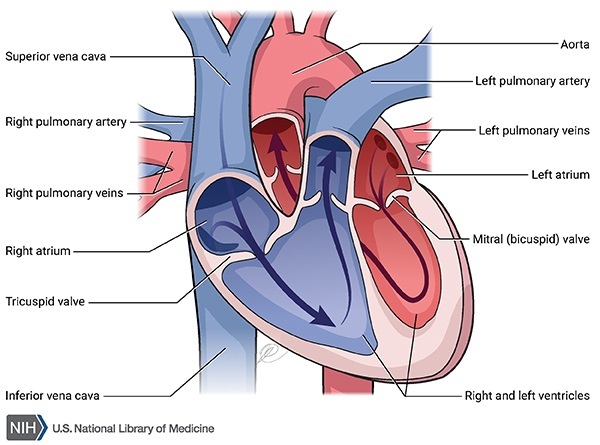
Individuals with liver GSD 0 usually show signs and symptoms of the disorder in infancy. People with this disorder develop low blood sugar (glucose), known as hypoglycemia, after going long periods of time without food (fasting). Signs of hypoglycemia become apparent when affected infants begin sleeping through the night and stop late-night feedings; these infants exhibit extreme tiredness (lethargy), pale skin (pallor), and nausea. During episodes of fasting, ketone levels in the blood may increase (ketosis). Ketones are molecules produced during the breakdown of fats, which occurs when stored sugars (such as glycogen) are unavailable. These short-term signs and symptoms of liver GSD 0 often improve when food is eaten and glucose levels in the body return to normal. The features of liver GSD 0 vary; they can be mild and go unnoticed for years, or they can include developmental delay and growth failure.
Frequency
The prevalence of GSD 0 is unknown; fewer than 10 people with the muscle type and fewer than 30 people with the liver type have been described in the scientific literature. Because some people with muscle GSD 0 die from sudden cardiac arrest early in life before a diagnosis is made and many with liver GSD 0 have mild signs and symptoms, it is thought that GSD 0 may be underdiagnosed.
Causes
Mutations in the GYS1 gene cause muscle GSD 0, and mutations in the GYS2 gene cause liver GSD 0. These genes provide instructions for making different versions of an enzyme called glycogen synthase. Both versions of glycogen synthase have the same function, to form glycogen molecules by linking together molecules of the simple sugar glucose, although they perform this function in different regions of the body.
The GYS1 gene provides instructions for making muscle glycogen synthase; this form of the enzyme is produced in most cells, but it is especially abundant in heart (cardiac) muscle and the muscles used for movement (skeletal muscles). During cardiac muscle contractions or rapid or sustained movement of skeletal muscle, glycogen stored in muscle cells is broken down to supply the cells with energy.
The GYS2 gene provides instructions for making liver glycogen synthase, which is produced solely in liver cells. Glycogen that is stored in the liver can be broken down rapidly when glucose is needed to maintain normal blood glucose levels between meals.
Mutations in the GYS1 or GYS2 gene lead to a lack of functional glycogen synthase, which prevents the production of glycogen from glucose. Mutations that cause GSD 0 result in a complete absence of glycogen in either liver or muscle cells. As a result, these cells do not have glycogen as a source of stored energy to draw upon following physical activity or fasting. This shortage of glycogen leads to the signs and symptoms of GSD 0.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Glycogen storage disease 0
- Glycogen synthase deficiency
- Glycogen synthetase deficiency
- GSD 0
- GSD type 0
- Hypoglycemia with deficiency of glycogen synthetase
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Bachrach BE, Weinstein DA, Orho-Melander M, Burgess A, Wolfsdorf JI. Glycogen synthase deficiency (glycogen storage disease type 0) presenting with hyperglycemia and glucosuria: report of three new mutations. J Pediatr. 2002 Jun;140(6):781-3. doi: 10.1067/mpd.2002.124317. Citation on PubMed
- Cameron JM, Levandovskiy V, MacKay N, Utgikar R, Ackerley C, Chiasson D, Halliday W, Raiman J, Robinson BH. Identification of a novel mutation in GYS1 (muscle-specific glycogen synthase) resulting in sudden cardiac death, that is diagnosable from skin fibroblasts. Mol Genet Metab. 2009 Dec;98(4):378-82. doi: 10.1016/j.ymgme.2009.07.012. Epub 2009 Jul 26. Citation on PubMed
- Fredriksson J, Anevski D, Almgren P, Sjogren M, Lyssenko V, Carlson J, Isomaa B, Taskinen MR, Groop L, Orho-Melander M; Botnia Study Group. Variation in GYS1 interacts with exercise and gender to predict cardiovascular mortality. PLoS One. 2007 Mar 14;2(3):e285. doi: 10.1371/journal.pone.0000285. Citation on PubMed or Free article on PubMed Central
- Groop L, Orho-Melander M. New insights into impaired muscle glycogen synthesis. PLoS Med. 2008 Jan 29;5(1):e25. doi: 10.1371/journal.pmed.0050025. Citation on PubMed or Free article on PubMed Central
- Kollberg G, Tulinius M, Gilljam T, Ostman-Smith I, Forsander G, Jotorp P, Oldfors A, Holme E. Cardiomyopathy and exercise intolerance in muscle glycogen storage disease 0. N Engl J Med. 2007 Oct 11;357(15):1507-14. doi: 10.1056/NEJMoa066691. Citation on PubMed
- Nessa A, Kumaran A, Kirk R, Dalton A, Ismail D, Hussain K. Mutational analysis of the GYS2 gene in patients diagnosed with ketotic hypoglycaemia. J Pediatr Endocrinol Metab. 2012;25(9-10):963-7. doi: 10.1515/jpem-2012-0165. Citation on PubMed
- Orho M, Bosshard NU, Buist NR, Gitzelmann R, Aynsley-Green A, Blumel P, Gannon MC, Nuttall FQ, Groop LC. Mutations in the liver glycogen synthase gene in children with hypoglycemia due to glycogen storage disease type 0. J Clin Invest. 1998 Aug 1;102(3):507-15. doi: 10.1172/JCI2890. Citation on PubMed or Free article on PubMed Central
- Soggia AP, Correa-Giannella ML, Fortes MA, Luna AM, Pereira MA. A novel mutation in the glycogen synthase 2 gene in a child with glycogen storage disease type 0. BMC Med Genet. 2010 Jan 5;11:3. doi: 10.1186/1471-2350-11-3. Citation on PubMed or Free article on PubMed Central
- Spiegel R, Mahamid J, Orho-Melander M, Miron D, Horovitz Y. The variable clinical phenotype of liver glycogen synthase deficiency. J Pediatr Endocrinol Metab. 2007 Dec;20(12):1339-42. doi: 10.1515/jpem.2007.20.12.1339. Citation on PubMed
- Sukigara S, Liang WC, Komaki H, Fukuda T, Miyamoto T, Saito T, Saito Y, Nakagawa E, Sugai K, Hayashi YK, Sugie H, Sasaki M, Nishino I. Muscle glycogen storage disease 0 presenting recurrent syncope with weakness and myalgia. Neuromuscul Disord. 2012 Feb;22(2):162-5. doi: 10.1016/j.nmd.2011.08.008. Epub 2011 Sep 29. Citation on PubMed
- Weinstein DA, Correia CE, Saunders AC, Wolfsdorf JI. Hepatic glycogen synthase deficiency: an infrequently recognized cause of ketotic hypoglycemia. Mol Genet Metab. 2006 Apr;87(4):284-8. doi: 10.1016/j.ymgme.2005.10.006. Epub 2005 Dec 6. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.