

Description
Gillespie syndrome is a disorder that involves eye abnormalities, weak muscle tone from birth (congenital hypotonia), problems with balance and coordinating movements (ataxia), and mild to moderate intellectual disability.
Gillespie syndrome is characterized by underdevelopment (hypoplasia) of the colored part of the eye (the iris). In most affected individuals, part of the iris is missing (partial aniridia) in both eyes. In addition, the irises have a characteristic uneven pattern known as "scalloping" at the inner (pupillary) edge. The pupils are enlarged (dilated) and are fixed, which means they do not get smaller (constrict) in response to light. These abnormalities are thought to result from problems in the development or maintenance of the tiny muscles that allow the pupil to contract (sphincter pupillae). The eye abnormalities can cause blurry vision (reduced visual acuity) and increased sensitivity to light (photophobia). Rapid, involuntary eye movements (nystagmus) can also occur in Gillespie syndrome.
The balance and movement problems in Gillespie syndrome result from hypoplasia of the cerebellum, which is the part of the brain that coordinates movement. This abnormality can cause hypotonia and delayed development of motor skills such as walking. In addition, difficulty controlling the muscles of the mouth can lead to delayed speech development. The difficulties with coordination generally become noticeable in early childhood when the individual is learning these skills. People with Gillespie syndrome usually continue to have an unsteady pattern of walking (gait) and speech problems throughout life.
Other features of Gillespie syndrome can include abnormalities in the bones of the spine (vertebrae) and malformations of the heart.
Frequency
The prevalence of Gillespie syndrome is unknown. Only a few dozen affected individuals have been described in the medical literature. It has been estimated that Gillespie syndrome accounts for about 2 percent of cases of aniridia.
Causes
Gillespie syndrome is caused by mutations in the ITPR1 gene. This gene provides instructions for making a protein that is part of a channel that controls the flow of positively charged calcium atoms (calcium ions) within cells. Four ITPR1 protein molecules join together in a complex (a homotetramer) to form the channel.
In response to certain signals, the ITPR1 channel releases calcium ions from storage in a cell structure called the endoplasmic reticulum into the surrounding cell fluid (the cytoplasm). Proper regulation of calcium ion concentration inside cells is important for the development and function of various tissues and organs.
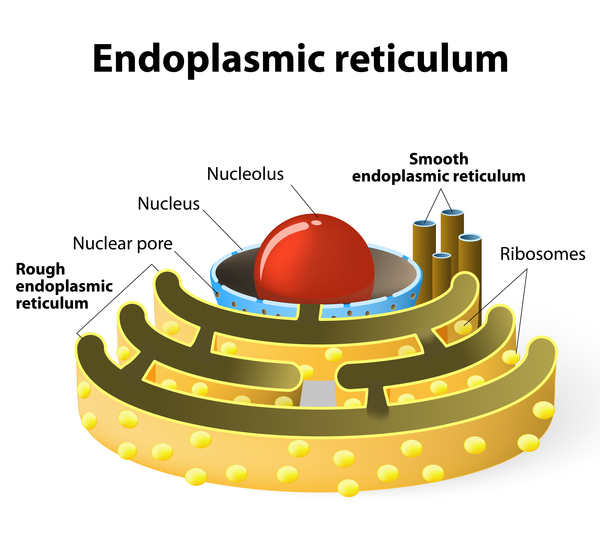
ITPR1 gene mutations likely result in a protein that cannot form stable calcium channels. A shortage of ITPR1 channels impairs the cell's ability to regulate the concentration of calcium ions. However, the specific connection between these changes and the signs and symptoms of Gillespie syndrome is unclear.
Inheritance
Gillespie syndrome can be inherited in an autosomal recessive pattern, which means both copies of a gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

In some cases of Gillespie syndrome, the condition occurs in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. Some affected individuals inherit the mutation from one affected parent. Other cases result from new mutations in the gene and occur in people with no history of the disorder in their family. Autosomal dominant cases of Gillespie syndrome are thought to arise from a dominant-negative effect, which means that the altered protein produced from one copy of the gene interferes with the function of the normal protein produced from the other copy of the gene.


Other Names for This Condition
- Aniridia, cerebellar ataxia, and mental retardation
- Aniridia-cerebellar ataxia-intellectual disability
- Aniridia-cerebellar ataxia-mental deficiency
- Partial aniridia-cerebellar ataxia-oligophrenia
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Dentici ML, Barresi S, Nardella M, Bellacchio E, Alfieri P, Bruselles A, Pantaleoni F, Danieli A, Iarossi G, Cappa M, Bertini E, Tartaglia M, Zanni G. Identification of novel and hotspot mutations in the channel domain of ITPR1 in two patients with Gillespie syndrome. Gene. 2017 Sep 10;628:141-145. doi: 10.1016/j.gene.2017.07.017. Epub 2017 Jul 8. Citation on PubMed or Free article on PubMed Central
- Gerber S, Alzayady KJ, Burglen L, Bremond-Gignac D, Marchesin V, Roche O, Rio M, Funalot B, Calmon R, Durr A, Gil-da-Silva-Lopes VL, Ribeiro Bittar MF, Orssaud C, Heron B, Ayoub E, Berquin P, Bahi-Buisson N, Bole C, Masson C, Munnich A, Simons M, Delous M, Dollfus H, Boddaert N, Lyonnet S, Kaplan J, Calvas P, Yule DI, Rozet JM, Fares Taie L. Recessive and Dominant De Novo ITPR1 Mutations Cause Gillespie Syndrome. Am J Hum Genet. 2016 May 5;98(5):971-980. doi: 10.1016/j.ajhg.2016.03.004. Epub 2016 Apr 21. Citation on PubMed or Free article on PubMed Central
- Hall HN, Williamson KA, FitzPatrick DR. The genetic architecture of aniridia and Gillespie syndrome. Hum Genet. 2019 Sep;138(8-9):881-898. doi: 10.1007/s00439-018-1934-8. Epub 2018 Sep 22. Citation on PubMed
- McEntagart M, Williamson KA, Rainger JK, Wheeler A, Seawright A, De Baere E, Verdin H, Bergendahl LT, Quigley A, Rainger J, Dixit A, Sarkar A, Lopez Laso E, Sanchez-Carpintero R, Barrio J, Bitoun P, Prescott T, Riise R, McKee S, Cook J, McKie L, Ceulemans B, Meire F, Temple IK, Prieur F, Williams J, Clouston P, Nemeth AH, Banka S, Bengani H, Handley M, Freyer E, Ross A; DDD Study; van Heyningen V, Marsh JA, Elmslie F, FitzPatrick DR. A Restricted Repertoire of De Novo Mutations in ITPR1 Cause Gillespie Syndrome with Evidence for Dominant-Negative Effect. Am J Hum Genet. 2016 May 5;98(5):981-992. doi: 10.1016/j.ajhg.2016.03.018. Epub 2016 Apr 21. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.