

Description
Fukuyama congenital muscular dystrophy is an inherited condition that predominantly affects the muscles , brain
, brain , and eyes
, and eyes . Congenital muscular dystrophies are a group of genetic conditions that cause muscle weakness and muscle wasting (atrophy) beginning early in life. The signs and symptoms of Fukuyama congenital muscular dystrophy can vary from mild to severe.
. Congenital muscular dystrophies are a group of genetic conditions that cause muscle weakness and muscle wasting (atrophy) beginning early in life. The signs and symptoms of Fukuyama congenital muscular dystrophy can vary from mild to severe.
Fukuyama congenital muscular dystrophy affects the skeletal muscles , which are the muscles the body uses for movement. The signs and symptoms of the disorder typically begin in early infancy and include a weak cry, difficulty feeding, and weak muscle tone (hypotonia). Weakness of the facial muscles often leads to a distinctive facial appearance including droopy eyelids (ptosis
, which are the muscles the body uses for movement. The signs and symptoms of the disorder typically begin in early infancy and include a weak cry, difficulty feeding, and weak muscle tone (hypotonia). Weakness of the facial muscles often leads to a distinctive facial appearance including droopy eyelids (ptosis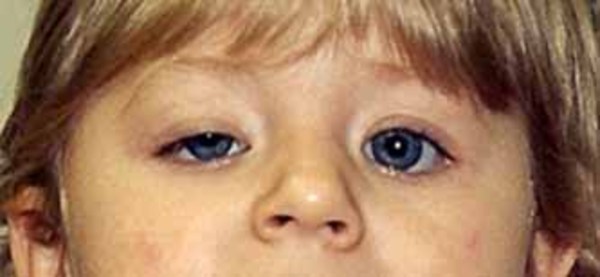 ) and an open mouth. In childhood, muscle weakness and joint deformities (contractures) restrict movement and interfere with the development of motor skills such as sitting, standing, and walking. Children with mild Fukuyama congenital muscular dystrophy may be able to stand or walk on their own, while those with severe signs and symptoms may not be able to sit without support.
) and an open mouth. In childhood, muscle weakness and joint deformities (contractures) restrict movement and interfere with the development of motor skills such as sitting, standing, and walking. Children with mild Fukuyama congenital muscular dystrophy may be able to stand or walk on their own, while those with severe signs and symptoms may not be able to sit without support.
Fukuyama congenital muscular dystrophy also impairs brain development. People with this condition often have a brain abnormality called cobblestone lissencephaly, in which the surface of the brain has a bumpy, irregular appearance (like that of cobblestones). These irregularities in the structure of the brain lead to delays in the development of motor skills and speech and moderate to severe intellectual disabilities. Social skills are less severely impaired. More than half of all affected children experience seizures.
In some people with Fukuyama congenital muscular dystrophy, vision is impaired. They may also have increased pressure in the eye (glaucoma) or abnormalities in the specialized light-sensitive tissue that lines the back of the eye (retina).
Individuals with Fukuyama congenital muscular dystrophy often develop heart problems in adolescence. These heart problems worsen over time. Severely affected individuals may also develop swallowing difficulties that can lead to a bacterial lung infection called aspiration pneumonia. The serious medical problems associated with Fukuyama congenital muscular dystrophy can shorten the life expectancy of someone with this condition.
Frequency
Fukuyama congenital muscular dystrophy is more common among people of Japanese ancestry. It is estimated to affect 1 in 25,000 to 50,000 infants born in Japan.
Causes
Fukuyama congenital muscular dystrophy is caused by variants (also called mutations) in the FKTN gene. This gene provides instructions for making an enzyme called fukutin. Fukutin modifies another protein called alpha-dystroglycan. This modification is necessary for alpha-dystroglycan to function. Alpha-dystroglycan anchors cells to the extracellular matrix, which is the network of molecules that forms in the spaces between cells and provides structural support. In skeletal muscles, alpha-dystroglycan helps stabilize and protect muscle fibers. In the brain, alpha-dystroglycan helps direct the movement (migration) of nerve cells (neurons ) during early development.
) during early development.
The most common variant in the FKTN gene reduces the amount of fukutin produced within cells. This impairs the cell's ability to produce functional alpha-dystroglycan. Without enough functional alpha-dystroglycan to stabilize muscle cells, muscle fibers become damaged as they repeatedly contract and relax with use. The damaged fibers weaken and die over time, leading to progressive weakness and atrophy of the skeletal muscles.
The lack of functional alpha-dystroglycan also impairs the migration of neurons during the early development of the brain. This leads to the cobblestone lissencephaly seen in children with Fukuyama congenital muscular dystrophy.
Because Fukuyama congenital muscular dystrophy involves problems with alpha-dystroglycan, the condition belongs to a group of disorders called dystroglycanopathies.
Inheritance
This condition is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
, which means both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
Other Names for This Condition
- Congenital muscular dystrophy, Fukuyama type
- FCMD
- FKTN-related congenital muscular dystrophy
- MDDGA4
- Muscular dystrophy-dystroglycanopathy (congenital with brain and eye anomalies), type A, 4
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Agarwal A, Sabat S, Kanekar S. Fukuyama Congenital Muscular Dystrophy. Cureus. 2022 Feb 4;14(2):e21902. doi: 10.7759/cureus.21902. eCollection 2022 Feb. Citation on PubMed
- Martin PT. Mechanisms of disease: congenital muscular dystrophies-glycosylation takes center stage. Nat Clin Pract Neurol. 2006 Apr;2(4):222-30. doi: 10.1038/ncpneuro0155. Citation on PubMed or Free article on PubMed Central
- Saito K. Fukuyama Congenital Muscular Dystrophy. 2006 Jan 26 [updated 2025 May 8]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1206/ Citation on PubMed
- Toda T, Kobayashi K, Takeda S, Sasaki J, Kurahashi H, Kano H, Tachikawa M, Wang F, Nagai Y, Taniguchi K, Taniguchi M, Sunada Y, Terashima T, Endo T, Matsumura K. Fukuyama-type congenital muscular dystrophy (FCMD) and alpha-dystroglycanopathy. Congenit Anom (Kyoto). 2003 Jun;43(2):97-104. doi: 10.1111/j.1741-4520.2003.tb01033.x. Citation on PubMed
- Yoshioka M, Higuchi Y, Fujii T, Aiba H, Toda T. Seizure-genotype relationship in Fukuyama-type congenital muscular dystrophy. Brain Dev. 2008 Jan;30(1):59-67. doi: 10.1016/j.braindev.2007.05.012. Epub 2007 Jun 26. Citation on PubMed
- Yoshioka M, Kuroki S. Clinical spectrum and genetic studies of Fukuyama congenital muscular dystrophy. Am J Med Genet. 1994 Nov 15;53(3):245-50. doi: 10.1002/ajmg.1320530309. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.