

Description
FG syndrome is a genetic condition that affects many parts of the body and occurs almost exclusively in males. "FG" represents the surname initials of the first family diagnosed with the disorder.
FG syndrome affects intelligence and behavior. Almost everyone with the condition has intellectual disability, which ranges from mild to severe. Affected individuals tend to be friendly, inquisitive, and hyperactive, with a short attention span. Compared to people with other forms of intellectual disability, their socialization and daily living skills are strong, while verbal communication and language skills tend to be weaker.
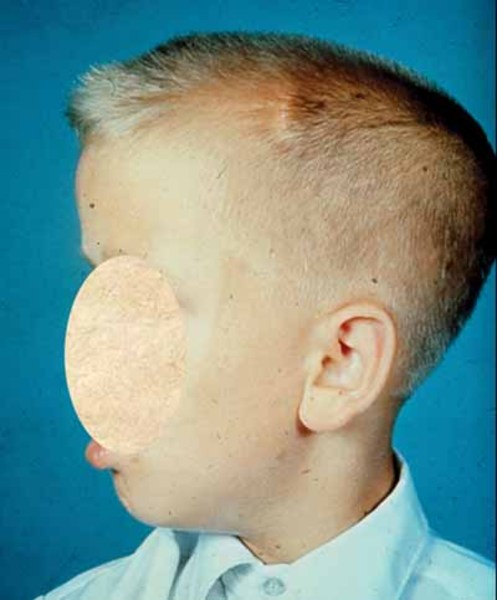
The physical features of FG syndrome include weak muscle tone (hypotonia), broad thumbs, and wide first (big) toes. Abnormalities of the tissue connecting the left and right halves of the brain (the corpus callosum) are also common. Most affected individuals have constipation, and many have abnormalities of the anus such as an obstruction of the anal opening (imperforate anus). People with FG syndrome also tend to have a distinctive facial appearance including small, underdeveloped ears; a tall, prominent forehead; and outside corners of the eyes that point downward (down-slanting palpebral fissures).

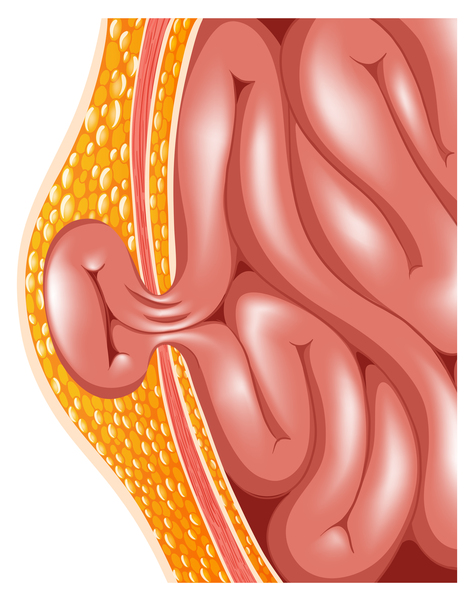
Additional features seen in some people with FG syndrome include widely set eyes (hypertelorism), an upswept frontal hairline, and a large head compared to body size (relative macrocephaly). Other health problems have also been reported, including heart defects, seizures, undescended testes (cryptorchidism) in males, and a soft out-pouching in the lower abdomen (an inguinal hernia).
Frequency
The prevalence of FG syndrome is unknown, although several hundred cases have been reported worldwide. Researchers suspect that FG syndrome may be overdiagnosed because many of its signs and symptoms are also seen with other disorders.
Causes
Researchers have identified changes in five regions of the X chromosome that are linked to FG syndrome in affected families. Mutations in a gene called MED12, which is located in one of these regions, appear to be the most common cause of the disorder. Researchers are investigating genes in other regions of the X chromosome that may also be associated with FG syndrome.
The MED12 gene provides instructions for making a protein that helps regulate gene activity. Specifically, the MED12 protein forms part of a large complex (a group of proteins that work together) that turns genes on and off. The MED12 protein is thought to play an essential role in development both before and after birth.
At least two mutations in the MED12 gene have been found to cause FG syndrome. Although the mutations alter the structure of the MED12 protein, it is unclear how they lead to intellectual disability, behavioral changes, and the physical features associated with this condition.
Inheritance
FG syndrome is inherited in an X-linked recessive pattern. The genes likely associated with this disorder, including MED12, are located on the X chromosome, which is one of the two sex chromosomes. In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a mutation usually must occur in both copies of the gene to cause the disorder. Because it is unlikely that females will have two altered copies of a gene on the X chromosome, males are affected by X-linked recessive disorders much more frequently than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.


Other Names for This Condition
- FGS
- FGS1
- Keller syndrome
- Mental retardation, large head, imperforate anus, congenital hypotonia, and partial agenesis of the corpus callosum
- OKS
- Opitz-Kaveggia syndrome
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Briault S, Hill R, Shrimpton A, Zhu D, Till M, Ronce N, Margaritte-Jeannin P, Baraitser M, Middleton-Price H, Malcolm S, Thompson E, Hoo J, Wilson G, Romano C, Guichet A, Pembrey M, Fontes M, Poustka A, Moraine C. A gene for FG syndrome maps in the Xq12-q21.31 region. Am J Med Genet. 1997 Nov 28;73(1):87-90. Citation on PubMed
- Briault S, Odent S, Lucas J, Le Merrer M, Turleau C, Munnich A, Moraine C. Paracentric inversion of the X chromosome [inv(X)(q12q28)] in familial FG syndrome. Am J Med Genet. 1999 Sep 10;86(2):112-4. doi: 10.1002/(sici)1096-8628(19990910)86:23.0.co;2-3. Citation on PubMed
- Clark RD, Graham JM Jr, Friez MJ, Hoo JJ, Jones KL, McKeown C, Moeschler JB, Raymond FL, Rogers RC, Schwartz CE, Battaglia A, Lyons MJ, Stevenson RE. FG syndrome, an X-linked multiple congenital anomaly syndrome: the clinical phenotype and an algorithm for diagnostic testing. Genet Med. 2009 Nov;11(11):769-75. doi: 10.1097/GIM.0b013e3181bd3d90. Citation on PubMed or Free article on PubMed Central
- Dessay S, Moizard MP, Gilardi JL, Opitz JM, Middleton-Price H, Pembrey M, Moraine C, Briault S. FG syndrome: linkage analysis in two families supporting a new gene localization at Xp22.3 [FGS3]. Am J Med Genet. 2002 Sep 15;112(1):6-11. doi: 10.1002/ajmg.10546. Citation on PubMed
- Graham JM Jr, Tackels D, Dibbern K, Superneau D, Rogers C, Corning K, Schwartz CE. FG syndrome: report of three new families with linkage to Xq12-q22.1. Am J Med Genet. 1998 Nov 2;80(2):145-56. Citation on PubMed
- Graham JM Jr, Visootsak J, Dykens E, Huddleston L, Clark RD, Jones KL, Moeschler JB, Opitz JM, Morford J, Simensen R, Rogers RC, Schwartz CE, Friez MJ, Stevenson RE. Behavior of 10 patients with FG syndrome (Opitz-Kaveggia syndrome) and the p.R961W mutation in the MED12 gene. Am J Med Genet A. 2008 Dec 1;146A(23):3011-7. doi: 10.1002/ajmg.a.32553. Citation on PubMed or Free article on PubMed Central
- Jehee FS, Rosenberg C, Krepischi-Santos AC, Kok F, Knijnenburg J, Froyen G, Vianna-Morgante AM, Opitz JM, Passos-Bueno MR. An Xq22.3 duplication detected by comparative genomic hybridization microarray (Array-CGH) defines a new locus (FGS5) for FG syndrome. Am J Med Genet A. 2005 Dec 15;139(3):221-6. doi: 10.1002/ajmg.a.30991. Citation on PubMed
- Lyons MJ, Graham JM Jr, Neri G, Hunter AG, Clark RD, Rogers RC, Moscarda M, Boccuto L, Simensen R, Dodd J, Robertson S, DuPont BR, Friez MJ, Schwartz CE, Stevenson RE. Clinical experience in the evaluation of 30 patients with a prior diagnosis of FG syndrome. J Med Genet. 2009 Jan;46(1):9-13. doi: 10.1136/jmg.2008.060509. Epub 2008 Sep 19. Citation on PubMed
- Lyons MJ. MED12-Related Disorders. 2008 Jun 23 [updated 2021 Aug 12]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1676/ Citation on PubMed
- Ozonoff S, Williams BJ, Rauch AM, Opitz JO. Behavior phenotype of FG syndrome: cognition, personality, and behavior in eleven affected boys. Am J Med Genet. 2000 Summer;97(2):112-8. doi: 10.1002/1096-8628(200022)97:23.0.co;2-d. Citation on PubMed
- Piluso G, Carella M, D'Avanzo M, Santinelli R, Carrano EM, D'Avanzo A, D'Adamo AP, Gasparini P, Nigro V. Genetic heterogeneity of FG syndrome: a fourth locus (FGS4) maps to Xp11.4-p11.3 in an Italian family. Hum Genet. 2003 Feb;112(2):124-30. doi: 10.1007/s00439-002-0863-7. Epub 2002 Nov 13. Citation on PubMed
- Risheg H, Graham JM Jr, Clark RD, Rogers RC, Opitz JM, Moeschler JB, Peiffer AP, May M, Joseph SM, Jones JR, Stevenson RE, Schwartz CE, Friez MJ. A recurrent mutation in MED12 leading to R961W causes Opitz-Kaveggia syndrome. Nat Genet. 2007 Apr;39(4):451-3. doi: 10.1038/ng1992. Epub 2007 Mar 4. Citation on PubMed
- Rump P, Niessen RC, Verbruggen KT, Brouwer OF, de Raad M, Hordijk R. A novel mutation in MED12 causes FG syndrome (Opitz-Kaveggia syndrome). Clin Genet. 2011 Feb;79(2):183-8. doi: 10.1111/j.1399-0004.2010.01449.x. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.