

Description
Familial lipoprotein lipase deficiency is an inherited condition that disrupts the normal breakdown of fats in the body, resulting in an increase of certain kinds of fats.
People with familial lipoprotein lipase deficiency typically develop signs and symptoms before age 10, with one-quarter showing symptoms by age 1. The first symptom of this condition is usually abdominal pain, which can vary from mild to severe. The abdominal pain is often due to inflammation of the pancreas (pancreatitis). These episodes of pancreatitis begin as sudden (acute) attacks. If left untreated, pancreatitis can develop into a chronic condition that can damage the pancreas and, in rare cases, be life-threatening.
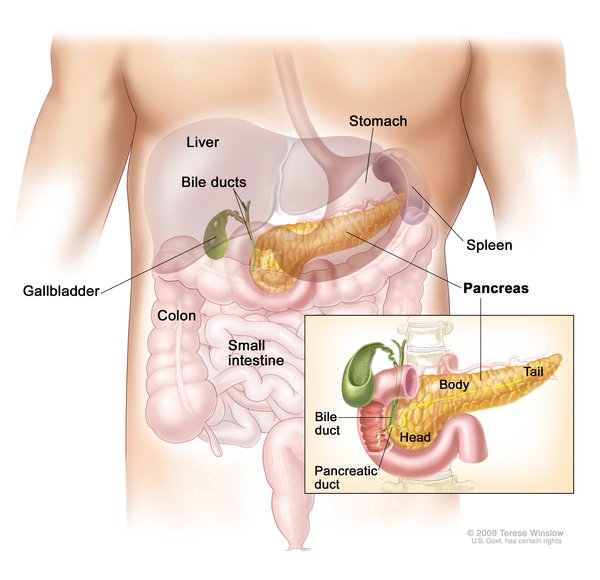
Affected individuals may also have an enlarged liver and spleen (hepatosplenomegaly). The higher the levels of fat in the body, the larger the liver and spleen become. As fat levels rise, certain white blood cells called macrophages take in excess fat in an attempt to rid fat from the bloodstream. After taking in fat, the macrophages travel to the liver and spleen, where the fatty cells accumulate.
Approximately half of individuals with familial lipoprotein lipase deficiency develop small yellow deposits of fat under the skin called eruptive xanthomas. These fat deposits most commonly appear on the trunk, buttocks, knees, and arms. Eruptive xanthomas are small (about 1 millimeter in diameter), but individual xanthomas can cluster together to form larger patches. They are generally not painful unless exposed to repeated friction or abrasion. Eruptive xanthomas begin to appear when fat intake increases and levels rise; the deposits disappear when fat intake slows and levels decrease.
The blood of people with familial lipoprotein lipase deficiency can have a milky appearance due to its high fat content. When fat levels get very high in people with this condition, fats can accumulate in blood vessels in the tissue that lines the back of the eye (the retina). The fat buildup gives this tissue a pale pink appearance when examined (lipemia retinalis). This fat accumulation does not affect vision and will disappear once fats from the diet are reduced and levels in the body decrease.

In people with familial lipoprotein lipase deficiency, increased fat levels can also cause neurological features, such as depression, memory loss, and mild intellectual decline (dementia). These problems are remedied when dietary fat levels normalize.
Frequency
This condition affects about 1 per million people worldwide. It is much more common in certain areas of the province of Quebec, Canada.
Causes
Mutations in the LPL gene cause familial lipoprotein lipase deficiency. The LPL gene provides instructions for producing an enzyme called lipoprotein lipase, which is found primarily on the surface of cells that line tiny blood vessels (capillaries) within muscles and fatty (adipose) tissue. This enzyme helps break down fats called triglycerides, which are carried by molecules called lipoproteins.
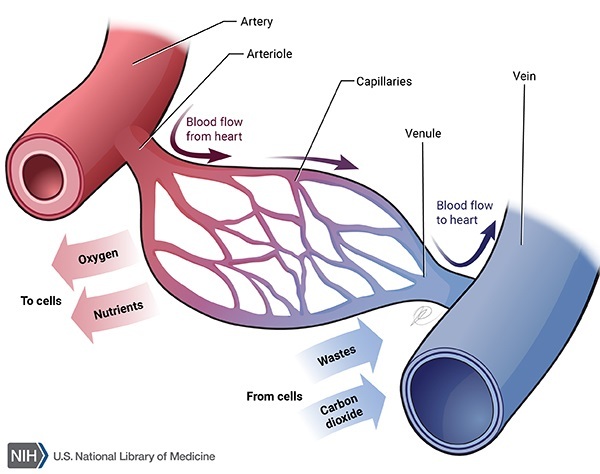
Mutations that cause familial lipoprotein lipase deficiency lead to a reduction or elimination of lipoprotein lipase activity, which prevents the enzyme from effectively breaking down triglycerides. As a result, triglycerides attached to lipoproteins build up in the blood and tissues, leading to the signs and symptoms of familial lipoprotein lipase deficiency.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Researchers speculate that if family members of affected individuals have a mutation in one copy of the LPL gene in each cell, they may have a mild increase in fat levels in the blood, which could increase their risk of health problems such as heart disease or diabetes. However, studies have not clearly demonstrated whether these individuals are more prone to develop these health problems than individuals in the general population.
Other Names for This Condition
- Burger-Grutz syndrome
- Endogenous hypertriglyceridaemia
- Familial fat-induced hypertriglyceridemia
- Familial hyperchylomicronemia
- Familial LPL deficiency
- Hyperlipoproteinemia type I
- Hyperlipoproteinemia type Ia
- Lipase D deficiency
- LIPD deficiency
- Lipoprotein lipase deficiency, familial
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Burnett JR, Hooper AJ, Hegele RA. Familial Lipoprotein Lipase Deficiency. 1999 Oct 12 [updated 2017 Jun 22]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1308/ Citation on PubMed
- Gilbert B, Rouis M, Griglio S, de Lumley L, Laplaud P. Lipoprotein lipase (LPL) deficiency: a new patient homozygote for the preponderant mutation Gly188Glu in the human LPL gene and review of reported mutations: 75 % are clustered in exons 5 and 6. Ann Genet. 2001 Jan-Mar;44(1):25-32. doi: 10.1016/s0003-3995(01)01037-1. Citation on PubMed
- Martin-Campos JM, Julve J, Roig R, Martinez S, Errico TL, Martinez-Couselo S, Escola-Gil JC, Mendez-Gonzalez J, Blanco-Vaca F. Molecular analysis of chylomicronemia in a clinical laboratory setting: diagnosis of 13 cases of lipoprotein lipase deficiency. Clin Chim Acta. 2014 Feb 15;429:61-8. doi: 10.1016/j.cca.2013.11.025. Epub 2013 Dec 1. Citation on PubMed
- Mohandas MK, Jemila J, Ajith Krishnan AS, George TT. Familial chylomicronemia syndrome. Indian J Pediatr. 2005 Feb;72(2):181. Citation on PubMed
- Sisman G, Erzin Y, Hatemi I, Caglar E, Boga S, Singh V, Senturk H. Familial chylomicronemia syndrome related chronic pancreatitis: a single-center study. Hepatobiliary Pancreat Dis Int. 2014 Apr;13(2):209-14. doi: 10.1016/s1499-3872(14)60033-3. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.