

Description
Familial hypobetalipoproteinemia (FHBL) is a disorder that impairs the body's ability to absorb and transport fats. This condition is characterized by low levels of a fat-like substance called cholesterol in the blood. The severity of signs and symptoms experienced by people with FHBL vary widely. The most mildly affected individuals have few problems with absorbing fats from the diet and no related signs and symptoms. Many individuals with FHBL develop an abnormal buildup of fats in the liver called hepatic steatosis or fatty liver. In more severely affected individuals, fatty liver may progress to chronic liver disease (cirrhosis). Individuals with severe FHBL have greater difficulty absorbing fats as well as fat-soluble vitamins such as vitamin E and vitamin A. This difficulty in fat absorption leads to excess fat in the feces (steatorrhea). In childhood, these digestive problems can result in an inability to grow or gain weight at the expected rate (failure to thrive).

Frequency
FHBL is estimated to occur in 1 in 1,000 to 3,000 individuals.
Causes
Most cases of FHBL are caused by mutations in the APOB gene. This gene provides instructions for making two versions of the apolipoprotein B protein: a short version called apolipoprotein B-48 and a longer version known as apolipoprotein B-100. Both of these proteins are components of lipoproteins, which transport fats and cholesterol in the blood.
Most APOB gene mutations that lead to FHBL cause both versions of apolipoprotein B to be abnormally short. The severity of the condition largely depends on the length of these two versions of apolipoprotein B. Severely shortened versions cannot partner with lipoproteins and transport fats and cholesterol. Proteins that are only slightly shortened retain some function but partner less effectively with lipoproteins. Generally, the signs and symptoms of FHBL are worse if both versions of apolipoprotein B are severely shortened. Mild or no signs and symptoms result when the proteins are only slightly shortened. All of these protein changes lead to a reduction of functional apolipoprotein B. As a result, the transportation of dietary fats and cholesterol is decreased or absent. A decrease in fat transport reduces the body's ability to absorb fats and fat-soluble vitamins from the diet.
Although APOB gene mutations are responsible for most cases of FHBL, mutations in a few other genes account for a small number of cases. Some people with FHBL do not have identified mutations in any of these genes. Changes in other, unidentified genes are likely involved in this condition.
Inheritance
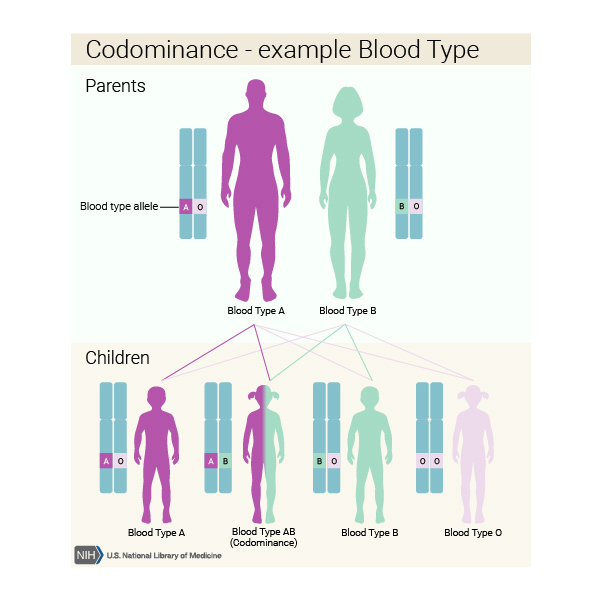
This condition is inherited in an autosomal codominant pattern. Codominance means that copies of the gene from both parents are active (expressed), and both copies influence the genetic trait. In FHBL, a change in one copy of the APOB gene in each cell can cause the condition, but changes in both copies of the gene cause more severe health problems.
Other Names for This Condition
- FHBL
- Hypobetalipoproteinemia
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Burnett JR, Hooper AJ, Hegele RA. APOB-Related Familial Hypobetalipoproteinemia. 2021 May 13 [updated 2021 Sep 9]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK570370/ Citation on PubMed
- Cariou B, Ouguerram K, Zair Y, Guerois R, Langhi C, Kourimate S, Benoit I, Le May C, Gayet C, Belabbas K, Dufernez F, Chetiveaux M, Tarugi P, Krempf M, Benlian P, Costet P. PCSK9 dominant negative mutant results in increased LDL catabolic rate and familial hypobetalipoproteinemia. Arterioscler Thromb Vasc Biol. 2009 Dec;29(12):2191-7. doi: 10.1161/ATVBAHA.109.194191. Epub 2009 Sep 17. Citation on PubMed
- Di Leo E, Magnolo L, Bertolotti M, Bourbon M, Carmo Pereira S, Pirisi M, Calandra S, Tarugi P. Variable phenotypic expression of homozygous familial hypobetalipoproteinaemia due to novel APOB gene mutations. Clin Genet. 2008 Sep;74(3):267-73. doi: 10.1111/j.1399-0004.2008.01023.x. Epub 2008 May 19. Citation on PubMed
- Fasano T, Cefalu AB, Di Leo E, Noto D, Pollaccia D, Bocchi L, Valenti V, Bonardi R, Guardamagna O, Averna M, Tarugi P. A novel loss of function mutation of PCSK9 gene in white subjects with low-plasma low-density lipoprotein cholesterol. Arterioscler Thromb Vasc Biol. 2007 Mar;27(3):677-81. doi: 10.1161/01.ATV.0000255311.26383.2f. Epub 2006 Dec 14. Citation on PubMed
- Hooper AJ, van Bockxmeer FM, Burnett JR. Monogenic hypocholesterolaemic lipid disorders and apolipoprotein B metabolism. Crit Rev Clin Lab Sci. 2005;42(5-6):515-45. doi: 10.1080/10408360500295113. Citation on PubMed
- Martin-Campos JM, Roig R, Mayoral C, Martinez S, Marti G, Arroyo JA, Julve J, Blanco-Vaca F. Identification of a novel mutation in the ANGPTL3 gene in two families diagnosed of familial hypobetalipoproteinemia without APOB mutation. Clin Chim Acta. 2012 Mar 22;413(5-6):552-5. doi: 10.1016/j.cca.2011.11.020. Epub 2011 Nov 29. Citation on PubMed
- Noto D, Cefalu AB, Valenti V, Fayer F, Pinotti E, Ditta M, Spina R, Vigna G, Yue P, Kathiresan S, Tarugi P, Averna MR. Prevalence of ANGPTL3 and APOB gene mutations in subjects with combined hypolipidemia. Arterioscler Thromb Vasc Biol. 2012 Mar;32(3):805-9. doi: 10.1161/ATVBAHA.111.238766. Epub 2012 Jan 12. Citation on PubMed
- Schonfeld G, Lin X, Yue P. Familial hypobetalipoproteinemia: genetics and metabolism. Cell Mol Life Sci. 2005 Jun;62(12):1372-8. doi: 10.1007/s00018-005-4473-0. Citation on PubMed
- Tarugi P, Averna M, Di Leo E, Cefalu AB, Noto D, Magnolo L, Cattin L, Bertolini S, Calandra S. Molecular diagnosis of hypobetalipoproteinemia: an ENID review. Atherosclerosis. 2007 Dec;195(2):e19-27. doi: 10.1016/j.atherosclerosis.2007.05.003. Epub 2007 Jun 14. Citation on PubMed
- Tarugi P, Averna M. Hypobetalipoproteinemia: genetics, biochemistry, and clinical spectrum. Adv Clin Chem. 2011;54:81-107. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.