

Description
The epilepsy-aphasia spectrum is a group of conditions that have overlapping signs and symptoms. A key feature of these conditions is impairment of language skills (aphasia). The language problems can affect speaking, reading, and writing. Another feature of epilepsy-aphasia spectrum disorders is certain patterns of abnormal electrical activity in the brain, which are detected by a test called an electroencephalogram (EEG). Many people with conditions in this spectrum develop recurrent seizures (epilepsy), and some have mild to severe intellectual disability. The conditions in the epilepsy-aphasia spectrum, which all begin in childhood, include Landau-Kleffner syndrome (LKS), epileptic encephalopathy with continuous spike-and-wave during sleep syndrome (ECSWS), autosomal dominant rolandic epilepsy with speech dyspraxia (ADRESD), intermediate epilepsy-aphasia disorder (IEAD), atypical childhood epilepsy with centrotemporal spikes (ACECTS), and childhood epilepsy with centrotemporal spikes (CECTS).
LKS and ECSWS are at the severe end of the spectrum. Both usually feature a characteristic abnormal pattern of electrical activity in the brain called continuous spike and waves during slow-wave sleep (CSWS). This pattern occurs while the affected child is sleeping, specifically during deep (slow-wave) sleep.
Most children with LKS develop normally in early childhood, although some speak later than their peers. However, affected children lose language skills beginning around age 5. This loss typically begins with verbal agnosia, which is the inability to understand speech. As LKS develops, the ability to express speech is also impaired. Approximately 70 percent of children with LKS have seizures, typically of a type described as focal (or partial) because the seizure activity occurs in specific regions of the brain rather than affecting the entire brain.
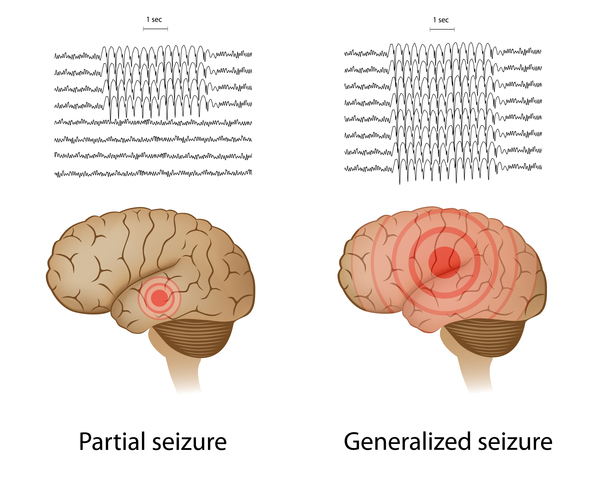
About half of children with ECSWS develop normally in early childhood, while others have delayed development of speech and motor skills. Although children with ECSWS typically lose a range of previously acquired skills, including those involved in language, movement, learning, or behavior, not everyone with ECSWS has aphasia. Seizures occur in approximately 80 percent of children with ECSWS and can include a variety of types, such as atypical absence seizures, which involve short periods of staring blankly; hemiclonic seizures, which cause rhythmic jerking of one side of the body; or generalized tonic-clonic seizures, which cause stiffening and rhythmic jerking of the entire body.
CECTS is at the mild end of the epilepsy-aphasia spectrum. Affected children have rolandic seizures; these seizures are triggered by abnormal activity in an area of the brain called the rolandic region, which is part of the cerebrum. The seizures, which usually occur during sleep, cause twitching, numbness, or tingling of the face or tongue, often causing drooling and impairing speech. In most people with CECTS, the seizures disappear by the end of adolescence. Most affected individuals develop normally, although some have difficulty coordinating the movements of the mouth and tongue needed for clear speech (dyspraxia) or impairment of language skills.
The other conditions in the epilepsy-aphasia spectrum are less common and fall in the middle of the spectrum. Children with IEAD usually have delayed development or regression of language skills. Some have seizures and most have abnormal electrical activity in their brains during sleep, although it is not prominent enough to be classified as CSWS. ACECTS features seizures and developmental regression that can affect movement, language, and attention. Children with ACECTS have abnormal electrical activity in the brain that is sometimes classified as CSWS. ADRESD is characterized by focal seizures, speech difficulties due to dyspraxia, and learning disability.
Frequency
The prevalence of the epilepsy-aphasia spectrum is unknown. Most of the conditions in the spectrum are rare; however, CECTS is one of the most common forms of epilepsy in children, accounting for 8 to 25 percent of cases. It is estimated to occur in 1 in 5,000 children younger than 16.
Causes
Variants (also known as mutations) in the GRIN2A gene can cause conditions in the epilepsy-aphasia spectrum. These variants are more common in the more severe conditions; they are found in up to 20 percent of people with LKS or ECSWS and about 5 percent of people with CECTS. In affected people without a GRIN2A gene variant, the cause of the condition is unknown. Some affected individuals have a brain abnormality that may contribute to the condition. Researchers suspect that changes in other, unidentified genes may also be associated with epilepsy-aphasia spectrum disorders.
The GRIN2A gene provides instructions for making the GluN2A protein, which is one component (subunit) of a subset of NMDA receptors. NMDA receptors transmit signals that turn on nerve cells (neurons ) in the brain. Signaling through these receptors is involved in normal brain development, changes in the brain in response to experience (synaptic plasticity), learning, and memory. The GluN2A subunit determines where in the brain the receptor is located and how it functions. Receptors containing this subunit are found in regions of the brain involved in speech and language, among other regions. These receptors also appear to play a role in brain signaling during slow-wave sleep.
) in the brain. Signaling through these receptors is involved in normal brain development, changes in the brain in response to experience (synaptic plasticity), learning, and memory. The GluN2A subunit determines where in the brain the receptor is located and how it functions. Receptors containing this subunit are found in regions of the brain involved in speech and language, among other regions. These receptors also appear to play a role in brain signaling during slow-wave sleep.
Variants in the GRIN2A gene lead to altered NMDA receptor signaling in the brain. As a result, neurons may be abnormally turned on, which can cause seizures and other abnormal brain activity and may lead to death of the neurons. Changes in GluN2A appear to particularly affect signaling in regions of the brain involved in speech and language and disrupt brain activity during slow-wave sleep, leading to several of the signs and symptoms of this group of conditions.
It is not clear why some people with a GRIN2A gene variant have a relatively mild condition and others have more severe signs and symptoms, even within the same family. Variations in other genes and environmental factors may also play a role in development of the condition.
Inheritance
Conditions in the epilepsy-aphasia spectrum that are caused by GRIN2A gene variants are inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. Individuals with an epilepsy-aphasia spectrum disorder may have family members with a condition in the epilepsy-aphasia spectrum or a related disorder such as isolated seizures or speech and language problems.
In some cases, an affected person inherits the variant from one affected parent . Other cases result from new variants in the gene
. Other cases result from new variants in the gene and occur in people with no history of the disorder in their family.
and occur in people with no history of the disorder in their family.
Other Names for This Condition
- Acquired aphasia with epilepsy
- DEE/EE-SWAS
- Developmental and/or epileptic encephalopathy with spike-wave activation in Sleep
- Epilepsy with continuous spike-wave in sleep
- Epilepsy with electrographic status epilepticus in sleep
- FESD
- Focal epilepsies with speech and language disorders
- Focal epilepsy with speech disorder and with or without mental retardation
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Carvill GL, Regan BM, Yendle SC, O'Roak BJ, Lozovaya N, Bruneau N, Burnashev N, Khan A, Cook J, Geraghty E, Sadleir LG, Turner SJ, Tsai MH, Webster R, Ouvrier R, Damiano JA, Berkovic SF, Shendure J, Hildebrand MS, Szepetowski P, Scheffer IE, Mefford HC. GRIN2A mutations cause epilepsy-aphasia spectrum disorders. Nat Genet. 2013 Sep;45(9):1073-6. doi: 10.1038/ng.2727. Epub 2013 Aug 11. Citation on PubMed or Free article on PubMed Central
- Lemke JR, Lal D, Reinthaler EM, Steiner I, Nothnagel M, Alber M, Geider K, Laube B, Schwake M, Finsterwalder K, Franke A, Schilhabel M, Jahn JA, Muhle H, Boor R, Van Paesschen W, Caraballo R, Fejerman N, Weckhuysen S, De Jonghe P, Larsen J, Moller RS, Hjalgrim H, Addis L, Tang S, Hughes E, Pal DK, Veri K, Vaher U, Talvik T, Dimova P, Guerrero Lopez R, Serratosa JM, Linnankivi T, Lehesjoki AE, Ruf S, Wolff M, Buerki S, Wohlrab G, Kroell J, Datta AN, Fiedler B, Kurlemann G, Kluger G, Hahn A, Haberlandt DE, Kutzer C, Sperner J, Becker F, Weber YG, Feucht M, Steinbock H, Neophythou B, Ronen GM, Gruber-Sedlmayr U, Geldner J, Harvey RJ, Hoffmann P, Herms S, Altmuller J, Toliat MR, Thiele H, Nurnberg P, Wilhelm C, Stephani U, Helbig I, Lerche H, Zimprich F, Neubauer BA, Biskup S, von Spiczak S. Mutations in GRIN2A cause idiopathic focal epilepsy with rolandic spikes. Nat Genet. 2013 Sep;45(9):1067-72. doi: 10.1038/ng.2728. Epub 2013 Aug 11. Citation on PubMed
- Lesca G, Rudolf G, Bruneau N, Lozovaya N, Labalme A, Boutry-Kryza N, Salmi M, Tsintsadze T, Addis L, Motte J, Wright S, Tsintsadze V, Michel A, Doummar D, Lascelles K, Strug L, Waters P, de Bellescize J, Vrielynck P, de Saint Martin A, Ville D, Ryvlin P, Arzimanoglou A, Hirsch E, Vincent A, Pal D, Burnashev N, Sanlaville D, Szepetowski P. GRIN2A mutations in acquired epileptic aphasia and related childhood focal epilepsies and encephalopathies with speech and language dysfunction. Nat Genet. 2013 Sep;45(9):1061-6. doi: 10.1038/ng.2726. Epub 2013 Aug 11. Citation on PubMed
- Specchio N, Wirrell EC, Scheffer IE, Nabbout R, Riney K, Samia P, Guerreiro M, Gwer S, Zuberi SM, Wilmshurst JM, Yozawitz E, Pressler R, Hirsch E, Wiebe S, Cross HJ, Perucca E, Moshe SL, Tinuper P, Auvin S. International League Against Epilepsy classification and definition of epilepsy syndromes with onset in childhood: Position paper by the ILAE Task Force on Nosology and Definitions. Epilepsia. 2022 Jun;63(6):1398-1442. doi: 10.1111/epi.17241. Epub 2022 May 3. Citation on PubMed
- Tsai MH, Vears DF, Turner SJ, Smith RL, Berkovic SF, Sadleir LG, Scheffer IE. Clinical genetic study of the epilepsy-aphasia spectrum. Epilepsia. 2013 Feb;54(2):280-7. doi: 10.1111/epi.12065. Epub 2013 Jan 7. Citation on PubMed
- Turner SJ, Morgan AT, Perez ER, Scheffer IE. New genes for focal epilepsies with speech and language disorders. Curr Neurol Neurosci Rep. 2015 Jun;15(6):35. doi: 10.1007/s11910-015-0554-0. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.