

Description
Epidermolysis bullosa with pyloric atresia (EB-PA) is a condition that affects the skin and digestive tract. This condition is one of several forms of epidermolysis bullosa, a group of genetic conditions that cause the skin to be fragile and to blister easily. Affected infants are often born with widespread blistering and areas of missing skin. Blisters continue to appear in response to minor injury or friction, such as rubbing or scratching. Most often, blisters occur over the whole body and affect mucous membranes such as the moist lining of the mouth and digestive tract.
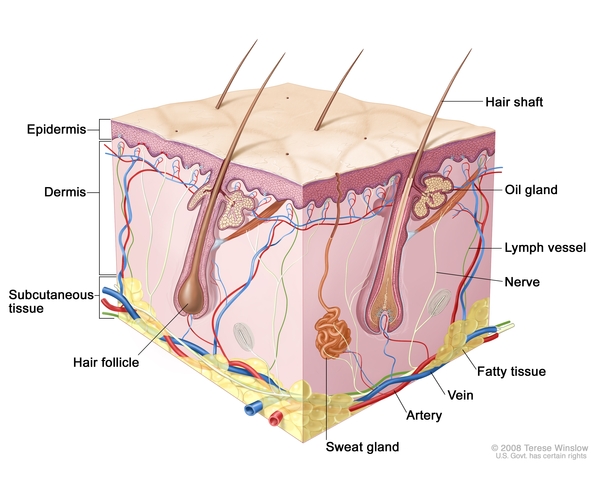
People with EB-PA are also born with pyloric atresia, which is a blockage (obstruction) of the lower part of the stomach (the pylorus). This obstruction prevents food from emptying out of the stomach into the intestine. Signs of pyloric atresia include vomiting, a swollen (distended) abdomen, and an absence of stool. Pyloric atresia is life-threatening and must be repaired with surgery soon after birth.
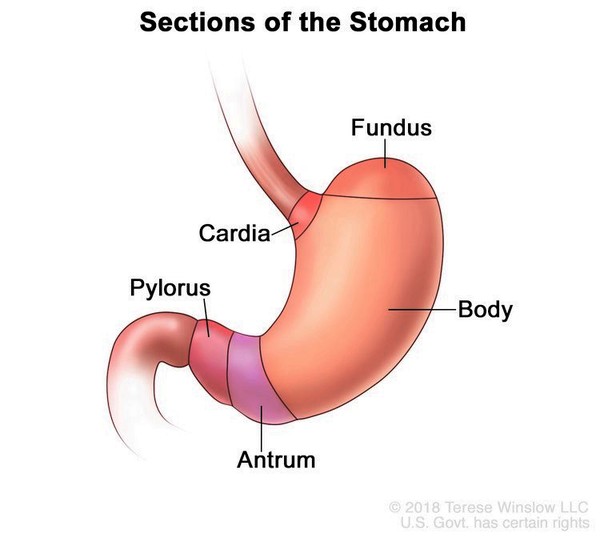
Other complications of EB-PA can include fusion of the skin between the fingers and toes, abnormalities of the fingernails and toenails, joint deformities (contractures) that restrict movement, and hair loss (alopecia). Some affected individuals are also born with malformations of the urinary tract, including the kidneys and bladder.

Because the signs and symptoms of EB-PA are so severe, many infants with this condition do not survive beyond the first year of life. In those who survive, the condition may improve with time; some affected individuals have little or no blistering later in life. However, many affected individuals who live past infancy experience severe health problems, including blistering and the formation of red, bumpy patches called granulation tissue. Granulation tissue most often forms on the skin around the mouth, nose, fingers, and toes. It can also build up in the airway, leading to difficulty breathing.
Frequency
EB-PA appears to be a rare condition, although its prevalence is unknown. At least 100 affected individuals have been reported worldwide.
Causes
EB-PA can be caused by mutations in the ITGA6, ITGB4, and PLEC genes. These genes provide instructions for making proteins with critical roles in the skin and digestive tract.
ITGB4 gene mutations are the most common cause of EB-PA; these mutations are responsible for about 80 percent of all cases. ITGA6 gene mutations cause about 5 percent of cases. The proteins produced from the ITGA6 and ITGB4 genes join to form a protein known as α6β4 integrin. This protein plays an important role in strengthening and stabilizing the skin by helping to attach the top layer of skin (the epidermis) to underlying layers. Mutations in either the ITGA6 gene or the ITGB4 gene lead to the production of a defective or nonfunctional version of α6β4 integrin, or prevent cells from making any of this protein. A shortage of functional α6β4 integrin causes cells in the epidermis to be fragile and easily damaged. Friction or other minor trauma can cause the skin layers to separate, leading to the formation of blisters.
About 15 percent of all cases of EB-PA result from mutations in the PLEC gene. This gene provides instructions for making a protein called plectin. Like α6β4 integrin, plectin helps attach the epidermis to underlying layers of skin. Some PLEC gene mutations prevent the cell from making any functional plectin, while other mutations result in an abnormal form of the protein. When plectin is altered or missing, the skin is less resistant to friction and minor trauma and blisters easily.
Researchers are working to determine how mutations in the ITGA6, ITGB4, and PLEC genes lead to pyloric atresia in people with EB-PA. Studies suggest that these genes are important for the normal development of the digestive tract.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Carmi syndrome
- EB-PA
- Junctional epidermolysis bullosa with pyloric atresia
- PA-JEB
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Lucky AW, Gorell E. Epidermolysis Bullosa with Pyloric Atresia. 2008 Feb 22 [updated 2023 Jan 26]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1157/ Citation on PubMed
- Pfendner E, Uitto J. Plectin gene mutations can cause epidermolysis bullosa with pyloric atresia. J Invest Dermatol. 2005 Jan;124(1):111-5. doi: 10.1111/j.0022-202X.2004.23564.x. Citation on PubMed
- Pfendner EG, Bruckner A, Conget P, Mellerio J, Palisson F, Lucky AW. Basic science of epidermolysis bullosa and diagnostic and molecular characterization: Proceedings of the IInd International Symposium on Epidermolysis Bullosa, Santiago, Chile, 2005. Int J Dermatol. 2007 Aug;46(8):781-94. doi: 10.1111/j.1365-4632.2007.03307.x. No abstract available. Citation on PubMed
- Pulkkinen L, Kim DU, Uitto J. Epidermolysis bullosa with pyloric atresia: novel mutations in the beta4 integrin gene (ITGB4). Am J Pathol. 1998 Jan;152(1):157-66. Citation on PubMed or Free article on PubMed Central
- Pulkkinen L, Kimonis VE, Xu Y, Spanou EN, McLean WH, Uitto J. Homozygous alpha6 integrin mutation in junctional epidermolysis bullosa with congenital duodenal atresia. Hum Mol Genet. 1997 May;6(5):669-74. doi: 10.1093/hmg/6.5.669. Citation on PubMed
- Pulkkinen L, Uitto J. Mutation analysis and molecular genetics of epidermolysis bullosa. Matrix Biol. 1999 Feb;18(1):29-42. doi: 10.1016/s0945-053x(98)00005-5. Citation on PubMed
- Varki R, Sadowski S, Pfendner E, Uitto J. Epidermolysis bullosa. I. Molecular genetics of the junctional and hemidesmosomal variants. J Med Genet. 2006 Aug;43(8):641-52. doi: 10.1136/jmg.2005.039685. Epub 2006 Feb 10. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.