

Description
Emanuel syndrome is a chromosomal disorder that disrupts normal development and affects many parts of the body. Infants with Emanuel syndrome have weak muscle tone (hypotonia) and fail to gain weight and grow at the expected rate (failure to thrive). Their development is significantly delayed, and most affected individuals have severe to profound intellectual disability.
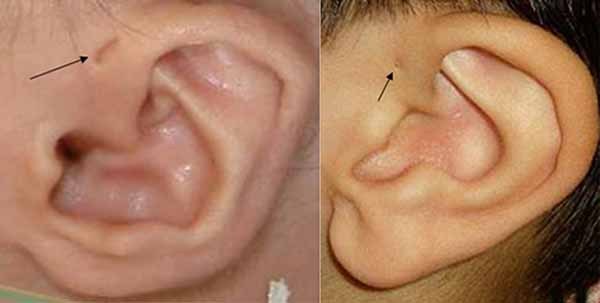
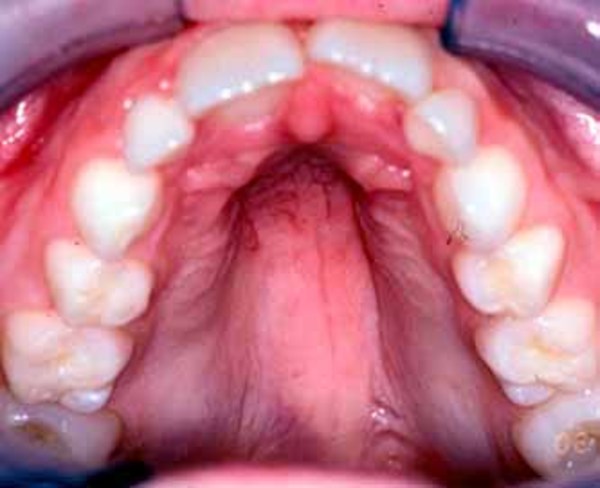
Other features of Emanuel syndrome include an unusually small head (microcephaly), distinctive facial features, and a small lower jaw (micrognathia). Ear abnormalities are common, including small holes in the skin just in front of the ears (preauricular pits or sinuses). About half of all affected infants are born with an opening in the roof of the mouth (cleft palate) or a high arched palate. Males with Emanuel syndrome often have genital abnormalities. Additional signs of this condition can include heart defects and absent or unusually small (hypoplastic) kidneys; these problems can be life-threatening in infancy or childhood.

Frequency
Emanuel syndrome is a rare disorder; its prevalence is unknown. More than 100 individuals with this condition have been reported.
Causes
Emanuel syndrome is caused by the presence of extra genetic material from chromosome 11 and chromosome 22 in each cell. In addition to the usual 46 chromosomes, people with Emanuel syndrome have an extra (supernumerary) chromosome consisting of a piece of chromosome 11 attached to a piece of chromosome 22. The extra chromosome is known as a derivative 22 or der(22) chromosome.
As a result of the extra chromosome, people with Emanuel syndrome have three copies of some genes in each cell instead of the usual two copies. The excess genetic material disrupts the normal course of development, leading to the characteristic signs and symptoms of this disorder. Researchers are working to determine which genes are included on the der(22) chromosome and what role these genes play in development.
Inheritance
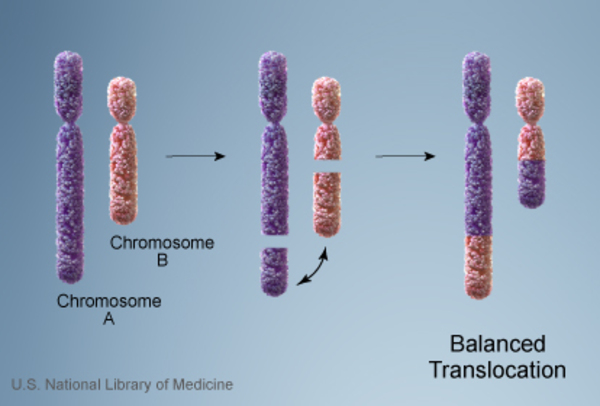
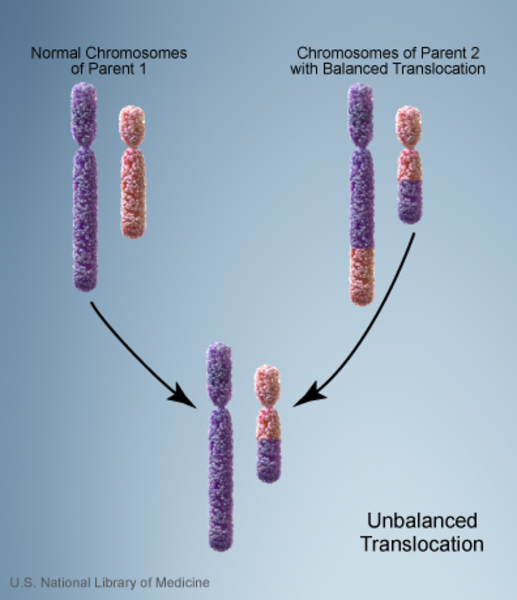
Almost everyone with Emanuel syndrome inherits the der(22) chromosome from an unaffected parent. The parent carries a chromosomal rearrangement between chromosomes 11 and 22 called a balanced translocation. No genetic material is gained or lost in a balanced translocation, so these chromosomal changes usually do not cause any health problems. However, translocations can become unbalanced as they are passed to the next generation. Individuals with Emanuel syndrome inherit an unbalanced translocation between chromosomes 11 and 22 that introduces extra genetic material in the form of the der(22) chromosome. This extra genetic material causes birth defects and the other health problems characteristic of this disorder.
Other Names for This Condition
- Der(22) syndrome due to 3:1 meiotic disjunction events
- Supernumerary der(22) syndrome
- Supernumerary der(22)t(11;22) syndrome
- Supernumerary derivative 22 chromosome syndrome
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Carter MT, St Pierre SA, Zackai EH, Emanuel BS, Boycott KM. Phenotypic delineation of Emanuel syndrome (supernumerary derivative 22 syndrome): Clinical features of 63 individuals. Am J Med Genet A. 2009 Aug;149A(8):1712-21. doi: 10.1002/ajmg.a.32957. Citation on PubMed or Free article on PubMed Central
- Emanuel BS, Zackai EH, Medne L. Emanuel Syndrome. 2007 Apr 20 [updated 2017 Aug 31]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1263/ Citation on PubMed
- Fraccaro M, Lindsten J, Ford CE, Iselius L. The 11q;22q translocation: a European collaborative analysis of 43 cases. Hum Genet. 1980;56(1):21-51. doi: 10.1007/BF00281567. No abstract available. Citation on PubMed
- Iselius L, Lindsten J, Aurias A, Fraccaro M, Bastard C, Bottelli AM, Bui TH, Caufin D, Dalpra L, Delendi N, et al. The 11q;22q translocation: a collaborative study of 20 new cases and analysis of 110 families. Hum Genet. 1983;64(4):343-55. doi: 10.1007/BF00292366. Citation on PubMed
- McDermid HE, Morrow BE. Genomic disorders on 22q11. Am J Hum Genet. 2002 May;70(5):1077-88. doi: 10.1086/340363. Epub 2002 Mar 29. Citation on PubMed or Free article on PubMed Central
- Prieto JC, Garcia NM, Elder FF, Zinn AR, Baker LA. Phenotypic expansion of the supernumerary derivative (22) chromosome syndrome: VACTERL and Hirschsprung's disease. J Pediatr Surg. 2007 Nov;42(11):1928-32. doi: 10.1016/j.jpedsurg.2007.07.030. Citation on PubMed
- Shaikh TH, Budarf ML, Celle L, Zackai EH, Emanuel BS. Clustered 11q23 and 22q11 breakpoints and 3:1 meiotic malsegregation in multiple unrelated t(11;22) families. Am J Hum Genet. 1999 Dec;65(6):1595-607. doi: 10.1086/302666. Citation on PubMed or Free article on PubMed Central
- Zackai EH, Emanuel BS. Site-specific reciprocal translocation, t(11;22) (q23;q11), in several unrelated families with 3:1 meiotic disjunction. Am J Med Genet. 1980;7(4):507-21. doi: 10.1002/ajmg.1320070412. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.