

Description
Dowling-Degos disease is a skin condition characterized by a lacy or net-like (reticulate) pattern of abnormally dark skin coloring (hyperpigmentation), particularly in the body's folds and creases. These skin changes typically first appear in the armpits and groin area and can later spread to other skin folds such as the crook of the elbow, back of the knee, and under the breasts. Less commonly, pigmentation changes can also occur on the neck, wrists, back of the hands, face, scalp, scrotum, and vulva. These areas of hyperpigmentation typically cause no health problems.
Individuals with Dowling-Degos disease may also have dark spots (lesions) on the face and back that resemble blackheads, red bumps around the mouth that resemble acne, or pitted scars on the face similar to acne scars but with no history of acne. Fluid-filled sacs within the hair follicle (pilar cysts) may develop, most commonly on the scalp. Rarely, affected individuals have patches of skin that are unusually light in color (hypopigmented).
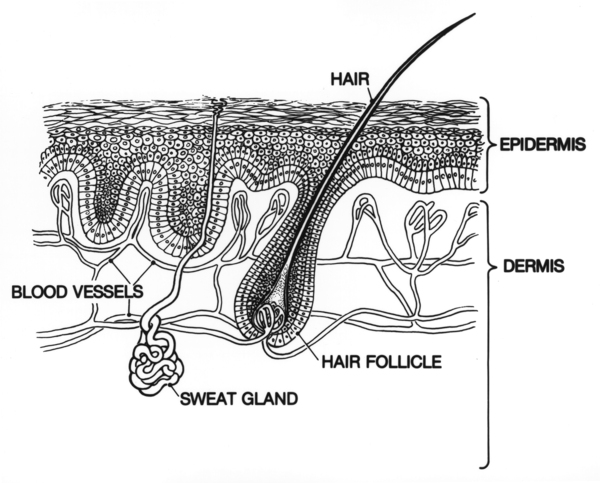
In rare cases, individuals with Dowling-Degos disease experience itching (pruritus) or burning sensations on the skin. These feelings can be triggered by UV light, sweating, or friction on the skin.
The pigmentation changes characteristic of Dowling-Degos disease typically begin in late childhood or in adolescence, although in some individuals, features of the condition do not appear until adulthood. New areas of hyperpigmentation tend to develop over time, and the other skin lesions tend to increase in number as well. While the skin changes associated with Dowling-Degos disease may cause distress or anxiety, they typically cause no other health problems.
A condition called Galli-Galli disease has signs and symptoms similar to those of Dowling-Degos disease. In addition to pigmentation changes, individuals with Galli-Galli disease also have a breakdown of cells in the outer layer of skin (acantholysis). Acantholysis can cause skin irritation and itchiness and lead to reddened or missing patches of skin (erosions). These conditions used to be considered two separate disorders, but Galli-Galli disease and Dowling-Degos disease are now regarded as the same condition.
Frequency
Dowling-Degos disease appears to be a rare condition, although its prevalence is unknown.
Causes
Mutations in the KRT5, POFUT1, or POGLUT1 gene cause most cases of Dowling-Degos disease.
The KRT5 gene provides instructions for making a protein called keratin 5, which is produced in cells called keratinocytes in the outer layer of the skin (the epidermis). Keratin 5 is one component of molecules called keratin intermediate filaments. These filaments assemble into strong networks that help attach (bind) keratinocytes together and anchor the epidermis to underlying layers of skin. Researchers believe that keratin 5 may also play a role in transporting melanosomes, which are pigment-carrying structures found in skin cells called melanocytes. The transport of these structures from melanocytes into keratinocytes is important for the development of normal skin coloration (pigmentation).
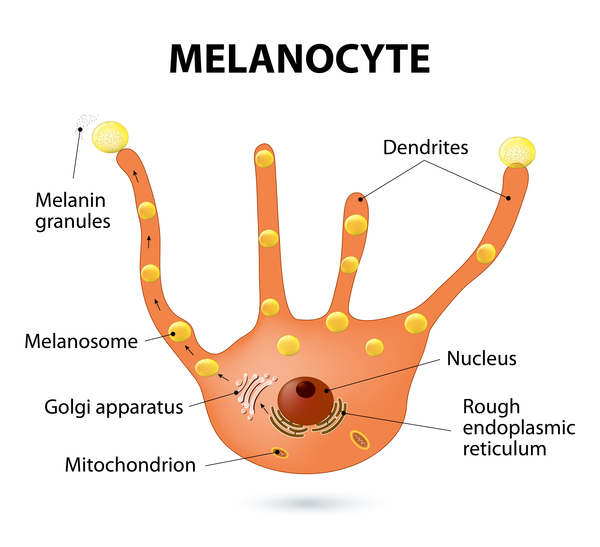
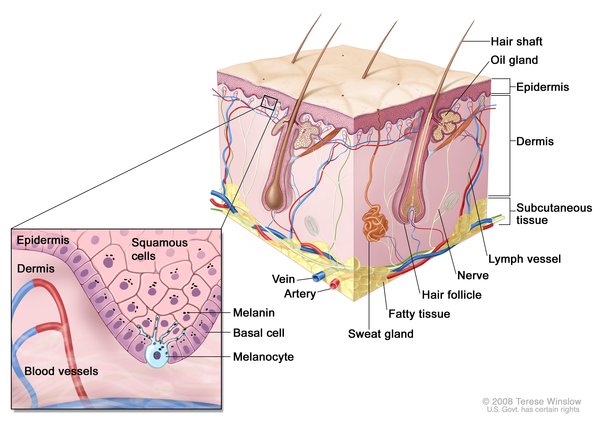
The POFUT1 and POGLUT1 genes provide instructions for making proteins that add different sugar molecules to proteins called Notch receptors. Notch receptors are a family of proteins that are involved in a signaling pathway that guides normal development of many tissues throughout the body, both before birth and throughout life. Receptor proteins have specific sites into which certain other proteins, called ligands, fit like keys into locks. The addition of sugar molecules to Notch receptors changes the shape of the receptors, allowing them to bind to their ligands and trigger signaling in the pathway. In skin cells, Notch signaling likely plays a role in maintaining precursor cells that mature into melanocytes and regulating interactions between melanocytes and keratinocytes.
KRT5 gene mutations that cause Dowling-Degos disease lead to a decrease in functional keratin 5 protein. A loss of keratin 5 can impair the formation of keratin intermediate filaments. As a result, the normal organization of the epidermis is altered, leading to the development of different types of skin lesions. Additionally, a decrease in keratin 5 may disrupt the transfer of pigment-carrying melanosomes from melanocytes to keratinocytes, where they are needed for the development of normal skin pigmentation. This disruption of melanosome transport is thought to cause the pigmentation abnormalities seen in individuals with Dowling-Degos disease.
Mutations in the POFUT1 or POGLUT1 gene result in a protein with little or no function. As a result, the protein is less able or unable to add sugar molecules to Notch receptors. Without these sugar molecules, Notch receptors cannot bind to their ligands and the Notch signaling pathway is halted. Because the varied functions of the Notch signaling pathway affect many body systems and Dowling-Degos disease affects only the skin, it is unclear whether the signs and symptoms of this condition are due to impaired Notch signaling or disruption of an unknown function of the protein in melanocytes or other skin cells.
Mutations in other genes, some of which have not been identified, are responsible for the remaining cases of Dowling-Degos disease.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.

Other Names for This Condition
- Dark dot disease
- DDD
- Dowling-Degos-Kitamura disease
- Reticular pigment anomaly of flexures
- Reticular pigmented anomaly of flexures
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Basmanav FB, Oprisoreanu AM, Pasternack SM, Thiele H, Fritz G, Wenzel J, Grosser L, Wehner M, Wolf S, Fagerberg C, Bygum A, Altmuller J, Rutten A, Parmentier L, El Shabrawi-Caelen L, Hafner C, Nurnberg P, Kruse R, Schoch S, Hanneken S, Betz RC. Mutations in POGLUT1, encoding protein O-glucosyltransferase 1, cause autosomal-dominant Dowling-Degos disease. Am J Hum Genet. 2014 Jan 2;94(1):135-43. doi: 10.1016/j.ajhg.2013.12.003. Citation on PubMed or Free article on PubMed Central
- Betz RC, Planko L, Eigelshoven S, Hanneken S, Pasternack SM, Bussow H, Van Den Bogaert K, Wenzel J, Braun-Falco M, Rutten A, Rogers MA, Ruzicka T, Nothen MM, Magin TM, Kruse R. Loss-of-function mutations in the keratin 5 gene lead to Dowling-Degos disease. Am J Hum Genet. 2006 Mar;78(3):510-9. doi: 10.1086/500850. Epub 2006 Jan 19. Citation on PubMed or Free article on PubMed Central
- Buket Basmanav F, Fritz G, Lestringant GG, Pachat D, Hoffjan S, Fischer J, Wehner M, Wolf S, Thiele H, Altmuller J, Pulimood SA, Rutten A, Kruse R, Hanneken S, Frank J, Danda S, Bygum A, Betz RC. Pathogenicity of POFUT1 in Dowling-Degos disease: additional mutations and clinical overlap with reticulate acropigmentation of kitamura. J Invest Dermatol. 2015 Feb;135(2):615-618. doi: 10.1038/jid.2014.406. Epub 2014 Sep 17. No abstract available. Citation on PubMed
- Hanneken S, Rutten A, Pasternack SM, Eigelshoven S, El Shabrawi-Caelen L, Wenzel J, Braun-Falco M, Ruzicka T, Nothen MM, Kruse R, Betz RC. Systematic mutation screening of KRT5 supports the hypothesis that Galli-Galli disease is a variant of Dowling-Degos disease. Br J Dermatol. 2010 Jul;163(1):197-200. doi: 10.1111/j.1365-2133.2010.09741.x. Epub 2010 Mar 5. Citation on PubMed
- Li M, Cheng R, Liang J, Yan H, Zhang H, Yang L, Li C, Jiao Q, Lu Z, He J, Ji J, Shen Z, Li C, Hao F, Yu H, Yao Z. Mutations in POFUT1, encoding protein O-fucosyltransferase 1, cause generalized Dowling-Degos disease. Am J Hum Genet. 2013 Jun 6;92(6):895-903. doi: 10.1016/j.ajhg.2013.04.022. Epub 2013 May 16. Citation on PubMed or Free article on PubMed Central
- Ralser DJ, Basmanav FB, Tafazzoli A, Wititsuwannakul J, Delker S, Danda S, Thiele H, Wolf S, Busch M, Pulimood SA, Altmuller J, Nurnberg P, Lacombe D, Hillen U, Wenzel J, Frank J, Odermatt B, Betz RC. Mutations in gamma-secretase subunit-encoding PSENEN underlie Dowling-Degos disease associated with acne inversa. J Clin Invest. 2017 Apr 3;127(4):1485-1490. doi: 10.1172/JCI90667. Epub 2017 Mar 13. Citation on PubMed or Free article on PubMed Central
- Verma S, Pasternack SM, Rutten A, Ruzicka T, Betz RC, Hanneken S. The First Report of KRT5 Mutation Underlying Acantholytic Dowling-Degos Disease with Mottled Hypopigmentation in an Indian Family. Indian J Dermatol. 2014 Sep;59(5):476-80. doi: 10.4103/0019-5154.139884. Citation on PubMed or Free article on PubMed Central
- Zhang J, Li M, Yao Z. Updated review of genetic reticulate pigmentary disorders. Br J Dermatol. 2017 Oct;177(4):945-959. doi: 10.1111/bjd.15575. Epub 2017 Sep 27. Citation on PubMed
- Zimmermann CC, Sforza D, Macedo PM, Azulay-Abulafia L, Alves Mde F, Carneiro SC. Dowling-Degos disease: classic clinical and histopathological presentation. An Bras Dermatol. 2011 Sep-Oct;86(5):979-82. doi: 10.1590/s0365-05962011000500016. English, Portuguese. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.