

Description
Dopa-responsive dystonia is a disorder that involves involuntary muscle contractions, tremors, and other uncontrolled movements (dystonia). The features of this condition range from mild to severe. This form of dystonia is called dopa-responsive dystonia because the signs and symptoms typically improve with sustained use of a medication known as L-Dopa.
Signs and symptoms of dopa-responsive dystonia usually appear during childhood, most commonly around age 6. The first signs of the condition are typically the development of inward- and upward-turning feet (clubfeet) and dystonia in the lower limbs. The dystonia spreads to the upper limbs over time; beginning in adolescence, the whole body is typically involved. Affected individuals may have unusual limb positioning and a lack of coordination when walking or running. Some people with this condition have sleep problems or episodes of depression more frequently than would normally be expected.

Over time, affected individuals often develop a group of movement abnormalities called parkinsonism. These abnormalities include unusually slow movement (bradykinesia), muscle rigidity, tremors, and an inability to hold the body upright and balanced (postural instability).
The movement difficulties associated with dopa-responsive dystonia usually worsen with age but stabilize around age 30. A characteristic feature of dopa-responsive dystonia is worsening of movement problems later in the day and an improvement of symptoms in the morning, after sleep (diurnal fluctuation).
Rarely, the movement problems associated with dopa-responsive dystonia do not appear until adulthood. In these adult-onset cases, parkinsonism usually develops before dystonia, and movement problems are slow to worsen and do not show diurnal fluctuations.
Frequency
Dopa-responsive dystonia is estimated to affect 1 per million people worldwide. However, the disorder is likely underdiagnosed because the condition may not be identified in people with mild symptoms, or it may be misdiagnosed in people who have symptoms similar to other movement disorders.
Causes
Mutations in the GCH1 gene are the most common cause of dopa-responsive dystonia. Less often, mutations in the TH or SPR gene cause this condition.
The GCH1 gene provides instructions for making an enzyme called GTP cyclohydrolase. This enzyme is involved in the first of three steps in the production of a molecule called tetrahydrobiopterin (BH4). The SPR gene, which provides instructions for making the sepiapterin reductase enzyme, is involved in the last step of tetrahydrobiopterin production. Tetrahydrobiopterin helps process several protein building blocks (amino acids), and is involved in the production of chemicals called neurotransmitters, which transmit signals between nerve cells in the brain. Specifically, tetrahydrobiopterin is involved in the production of two neurotransmitters called dopamine and serotonin. Among their many functions, dopamine transmits signals within the brain to produce smooth physical movements, and serotonin regulates mood, emotion, sleep, and appetite.

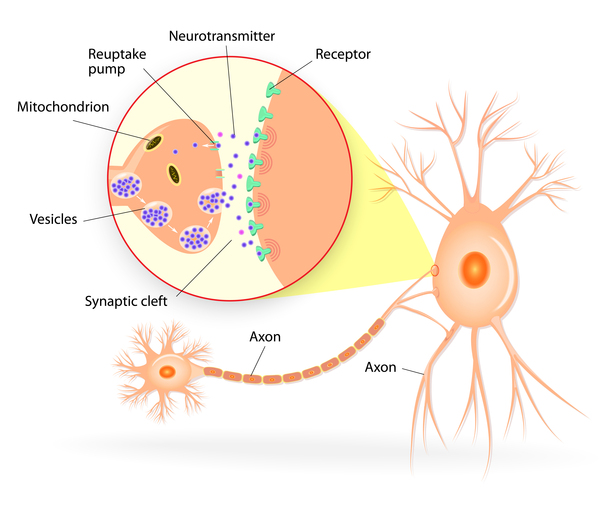
The protein produced from the TH gene is also involved in dopamine production. The TH gene provides instructions for making the enzyme tyrosine hydroxylase, which helps convert the amino acid tyrosine to dopamine.
Mutations in the GCH1 or SPR gene impair the production of tetrahydrobiopterin, which leads to a decrease in the amount of available dopamine. TH gene mutations result in the production of a tyrosine hydroxylase enzyme with reduced function, which leads to a decrease in dopamine production. A reduction in the amount of dopamine interferes with the brain's ability to produce smooth physical movements, resulting in the dystonia, tremor, and other movement problems associated with dopa-responsive dystonia. Sleep and mood disorders also occur in some individuals with GCH1 or SPR gene mutations; these disorders likely result from a disruption in the production of serotonin. Problems with sleep and episodes of depression are not seen in people with dopa-responsive dystonia caused by TH gene mutations, which is sometimes referred to as Segawa syndrome.
Some people with dopa-responsive dystonia do not have an identified mutation in the GCH1, TH, or SPR gene. The cause of the condition in these individuals is unknown.
Inheritance
When dopa-responsive dystonia is caused by mutations in the GCH1 gene, it is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. In some cases, an affected person inherits the mutation from one affected parent. Other cases result from new mutations in the gene and occur in people with no history of the disorder in their family.


Some people who inherit the altered GCH1 gene never develop features of dopa-responsive dystonia. (This situation is known as reduced penetrance.) It is unclear why some people with a mutated gene develop the disease and other people with a mutated gene do not. For unknown reasons, dopa-responsive dystonia caused by mutations in the GCH1 gene affects females two to four times more often than males.
When TH gene mutations are responsible for causing dopa-responsive dystonia, it is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

When dopa-responsive dystonia is caused by mutations in the SPR gene, it can have either an autosomal recessive or, less commonly, an autosomal dominant pattern of inheritance.
Other Names for This Condition
- DRD
- Dystonia 5, dopa-responsive type
- Hereditary progressive dystonia with marked diurnal fluctuation
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Arrabal L, Teresa L, Sanchez-Alcudia R, Castro M, Medrano C, Gutierrez-Solana L, Roldan S, Ormazabal A, Perez-Cerda C, Merinero B, Perez B, Artuch R, Ugarte M, Desviat LR. Genotype-phenotype correlations in sepiapterin reductase deficiency. A splicing defect accounts for a new phenotypic variant. Neurogenetics. 2011 Aug;12(3):183-91. doi: 10.1007/s10048-011-0279-4. Epub 2011 Mar 24. Citation on PubMed
- Asmus F, Gasser T. Dystonia-plus syndromes. Eur J Neurol. 2010 Jul;17 Suppl 1:37-45. doi: 10.1111/j.1468-1331.2010.03049.x. Citation on PubMed
- Bonafe L, Thony B, Penzien JM, Czarnecki B, Blau N. Mutations in the sepiapterin reductase gene cause a novel tetrahydrobiopterin-dependent monoamine-neurotransmitter deficiency without hyperphenylalaninemia. Am J Hum Genet. 2001 Aug;69(2):269-77. doi: 10.1086/321970. Epub 2001 Jul 6. Citation on PubMed or Free article on PubMed Central
- Schiller A, Wevers RA, Steenbergen GC, Blau N, Jung HH. Long-term course of L-dopa-responsive dystonia caused by tyrosine hydroxylase deficiency. Neurology. 2004 Oct 26;63(8):1524-6. doi: 10.1212/01.wnl.0000142083.47927.0a. Citation on PubMed
- Segawa M. Autosomal dominant GTP cyclohydrolase I (AD GCH 1) deficiency (Segawa disease, dystonia 5; DYT 5). Chang Gung Med J. 2009 Jan-Feb;32(1):1-11. Citation on PubMed
- Stafford BM, Klein E, Haskins K, Liebman E, Teslow T. Depth studies: illustrated anatomies from Vesalius to Vicq d'Azyr. Caduceus. 1992 Autumn;8(2):39-48. No abstract available. Citation on PubMed
- Steinberger D, Blau N, Goriuonov D, Bitsch J, Zuker M, Hummel S, Muller U. Heterozygous mutation in 5'-untranslated region of sepiapterin reductase gene (SPR) in a patient with dopa-responsive dystonia. Neurogenetics. 2004 Sep;5(3):187-90. doi: 10.1007/s10048-004-0182-3. Epub 2004 Jul 6. Citation on PubMed
- Trender-Gerhard I, Sweeney MG, Schwingenschuh P, Mir P, Edwards MJ, Gerhard A, Polke JM, Hanna MG, Davis MB, Wood NW, Bhatia KP. Autosomal-dominant GTPCH1-deficient DRD: clinical characteristics and long-term outcome of 34 patients. J Neurol Neurosurg Psychiatry. 2009 Aug;80(8):839-45. doi: 10.1136/jnnp.2008.155861. Epub 2009 Mar 29. Citation on PubMed
- Wu ZY, Lin Y, Chen WJ, Zhao GX, Xie H, Murong SX, Wang N. Molecular analyses of GCH-1, TH and parkin genes in Chinese dopa-responsive dystonia families. Clin Genet. 2008 Dec;74(6):513-21. doi: 10.1111/j.1399-0004.2008.01039.x. Epub 2008 Jun 11. Citation on PubMed
- Yeung WL, Wong VC, Chan KY, Hui J, Fung CW, Yau E, Ko CH, Lam CW, Mak CM, Siu S, Low L. Expanding phenotype and clinical analysis of tyrosine hydroxylase deficiency. J Child Neurol. 2011 Feb;26(2):179-87. doi: 10.1177/0883073810377014. Epub 2010 Sep 7. Citation on PubMed
- Zirn B, Steinberger D, Troidl C, Brockmann K, von der Hagen M, Feiner C, Henke L, Muller U. Frequency of GCH1 deletions in Dopa-responsive dystonia. J Neurol Neurosurg Psychiatry. 2008 Feb;79(2):183-6. doi: 10.1136/jnnp.2007.128413. Epub 2007 Sep 26. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.