

Description
Distal hereditary motor neuropathy, type II is a progressive disorder that affects nerve cells (neurons) in the brain and spinal cord. This condition specifically affects motor neurons, which are specialized cells that control muscle movement. Damage to motor neurons results in muscle weakness that worsens over time. Distal hereditary motor neuropathy, type II weakness primarily affects movement in the legs.

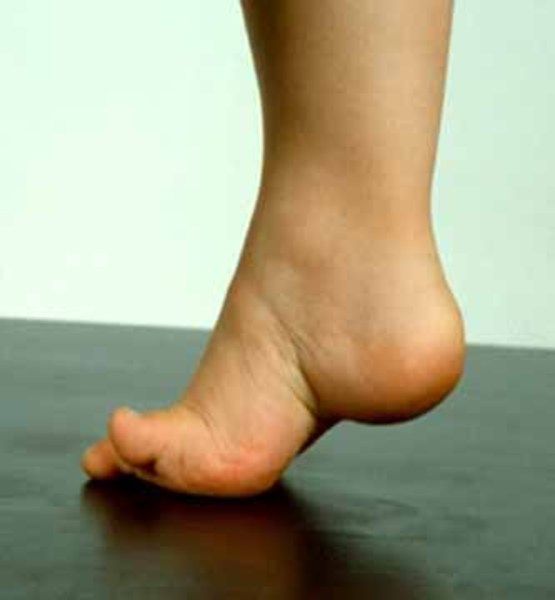
The signs and symptoms of distal hereditary motor neuropathy, type II often begin in adolescence to mid-adulthood. The initial symptoms of the disorder are cramps or weakness in the muscles of the big toe and, later, the entire foot. During the next 5 to 10 years, affected individuals experience a gradual loss of muscle tissue (atrophy) in the lower legs, which can lead to problems with walking (gait disturbance) and high arches (pes cavus). Over time, the lower legs may become paralyzed. The thigh muscles may also undergo muscle atrophy, although this generally occurs later and is less severe than the muscle atrophy in the lower legs.
Some individuals with distal hereditary motor neuropathy, type II can also experience weaken the muscles in the hands and forearms. This weakening is less severe than the weakening in the lower limbs and does not usually lead to paralysis. In rare cases, affected individuals experience hearing loss.
People with distal hereditary motor neuropathy, type II can have exaggerated reflexes (hyperreflexia) or other minor disturbances in the nerves used to detect sensations (sensory neuropathy). Sensory neuropathy is uncommon in people with distal hereditary motor neuropathy, type II and is typically a feature of a disorder called Charcot-Marie-Tooth disease. These two disorders have overlapping features and can also share a genetic
Frequency
The prevalence of all types of distal hereditary motor neuropathy is estimated to be about 2 in 100,000 individuals. Distal hereditary motor neuropathy, type II accounts for 8 to 15 percent of all cases of distal hereditary motor neuropathy.
Causes
Variants (also called mutations) in multiple genes can cause distal hereditary motor neuropathy, type II. Most commonly, variants in the HSPB1 and HSPB8 genes cause this condition. These genes provide instructions for making proteins called heat shock proteins. Heat shock protein beta-1 is made from the HSPB1 gene, and heat shock protein beta-8 is made from the HSPB8 gene. Heat shock proteins help protect cells that are under stress from factors such as infection, inflammation, exposure to toxins, elevated temperature, injury, and disease. In addition, these proteins appear to be involved in stabilizing the cell's structural framework (the cytoskeleton), folding and stabilizing newly produced proteins, and repairing damaged proteins. Heat shock proteins also seem to play a role in the tensing of muscle fibers (muscle contraction).
Heat shock protein beta-1 and heat shock protein beta-8 are found in cells throughout the body. These proteins help fold newly produced proteins and refold damaged proteins. These proteins are also involved in maintaining the structure of neurons.
The HSPB1 and HSPB8 gene variants that cause distal hereditary motor neuropathy, type II typically lead to changes in single protein building blocks (amino acids) in the proteins. These changes can cause the proteins to form clusters (aggregates). These aggregates can build up and impair the function of cells, particularly motor neurons. In addition, neurons that do not have functional versions of these heat shock proteins are vulnerable to stress and cell damage. As a result, these cells are more likely to die off over time, leading to the signs and symptoms of distal hereditary motor neuropathy, type II.
In rare cases, variants in other genes can cause distal hereditary motor neuropathy, type II.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.

Other Names for This Condition
- Distal hereditary motor neuronopathy, type II
- HMN II
- HMN2
- HMND2
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Benquey T, Pion E, Cossee M, Krahn M, Stojkovic T, Perrin A, Cerino M, Molon A, Lia AS, Magdelaine C, Francou B, Guiochon-Mantel A, Malinge MC, Leguern E, Levy N, Attarian S, Latour P, Bonello-Palot N. A National French Consensus on Gene List for the Diagnosis of Charcot-Marie-Tooth Disease and Related Disorders Using Next-Generation Sequencing. Genes (Basel). 2022 Feb 9;13(2):318. doi: 10.3390/genes13020318. Citation on PubMed
- Frasquet M, Rojas-Garcia R, Argente-Escrig H, Vazquez-Costa JF, Muelas N, Vilchez JJ, Sivera R, Millet E, Barreiro M, Diaz-Manera J, Turon-Sans J, Cortes-Vicente E, Querol L, Ramirez-Jimenez L, Martinez-Rubio D, Sanchez-Monteagudo A, Espinos C, Sevilla T, Lupo V. Distal hereditary motor neuropathies: Mutation spectrum and genotype-phenotype correlation. Eur J Neurol. 2021 Apr;28(4):1334-1343. doi: 10.1111/ene.14700. Epub 2021 Jan 10. Citation on PubMed
- Hu Z, Chen L, Zhang J, Li T, Tang J, Xu N, Wang X. Structure, function, property, and role in neurologic diseases and other diseases of the sHsp22. J Neurosci Res. 2007 Aug 1;85(10):2071-9. doi: 10.1002/jnr.21231. Citation on PubMed
- Irobi J, Van Impe K, Seeman P, Jordanova A, Dierick I, Verpoorten N, Michalik A, De Vriendt E, Jacobs A, Van Gerwen V, Vennekens K, Mazanec R, Tournev I, Hilton-Jones D, Talbot K, Kremensky I, Van Den Bosch L, Robberecht W, Van Vandekerckhove J, Van Broeckhoven C, Gettemans J, De Jonghe P, Timmerman V. Hot-spot residue in small heat-shock protein 22 causes distal motor neuropathy. Nat Genet. 2004 Jun;36(6):597-601. doi: 10.1038/ng1328. Epub 2004 May 2. Citation on PubMed
- Shen X, Zhang J, Zhan F, Tian W, Jiang Q, Luan X, Zhang X, Cao L. Heterogeneous Clinical Phenotypes of dHMN Caused by Mutation in HSPB1 Gene: A Case Series. Biomolecules. 2022 Sep 27;12(10):1382. doi: 10.3390/biom12101382. Citation on PubMed
- Tazir M, Nouioua S. Distal hereditary motor neuropathies. Rev Neurol (Paris). 2024 Dec;180(10):1031-1036. doi: 10.1016/j.neurol.2023.09.005. Epub 2024 May 3. Citation on PubMed
- Tedesco B, Cristofani R, Ferrari V, Cozzi M, Rusmini P, Casarotto E, Chierichetti M, Mina F, Galbiati M, Piccolella M, Crippa V, Poletti A. Insights on Human Small Heat Shock Proteins and Their Alterations in Diseases. Front Mol Biosci. 2022 Feb 25;9:842149. doi: 10.3389/fmolb.2022.842149. eCollection 2022. Citation on PubMed
- Xie Y, Lin Z, Pakhrin PS, Li X, Wang B, Liu L, Huang S, Zhao H, Cao W, Hu Z, Guo J, Shen L, Tang B, Zhang R. Genetic and Clinical Features in 24 Chinese Distal Hereditary Motor Neuropathy Families. Front Neurol. 2020 Dec 14;11:603003. doi: 10.3389/fneur.2020.603003. eCollection 2020. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.