

Description
Congenital plasminogen deficiency is a disorder that results in inflamed growths on the mucous membranes, which are the moist tissues that line body openings such as the eyelids and the inside of the mouth. Development of the growths are usually triggered by infections or injury, but they may also occur spontaneously in the absence of known triggers. The growths may recur after being removed.
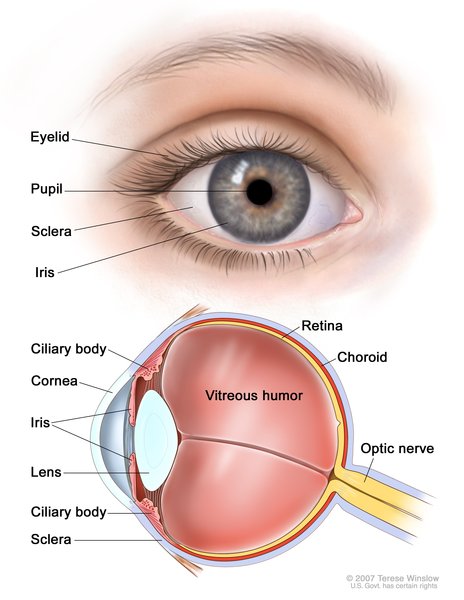
Congenital plasminogen deficiency most often affects the conjunctiva, which are the mucous membranes that protect the white part of the eye (the sclera) and line the eyelids. A characteristic feature of this disorder is ligneous conjunctivitis, in which a buildup of a protein called fibrin causes inflammation of the conjunctiva (conjunctivitis) and leads to thick, woody (ligneous), inflamed growths that are yellow, white, or red. Ligneous conjunctivitis most often occurs on the inside of the eyelids. However, in about one-third of cases, ligneous conjunctivitis over the sclera grows onto the cornea, which is the clear covering that protects the colored part of the eye (the iris) and pupil. Such growths can tear the cornea or cause scarring. These corneal problems as well as obstruction by growths inside the eyelid can lead to vision loss.
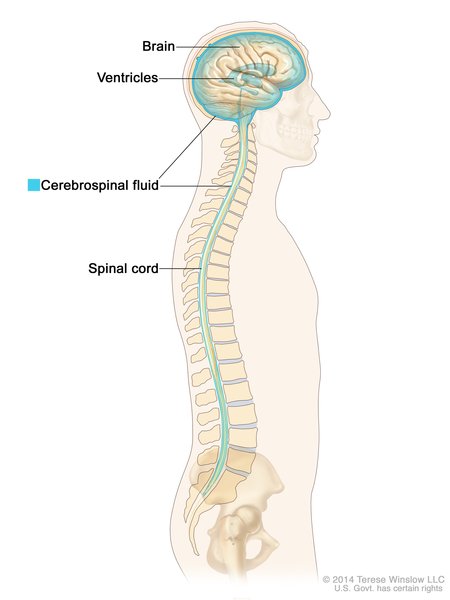
People with congenital plasminogen deficiency may also develop ligneous growths on other mucous membranes, including the inside of the mouth and the gums; the lining of the nasal cavity; and in females, the vagina. Growths on the mucous membranes that line the gastrointestinal tract may result in ulcers. The growths may also develop in the windpipe, which can cause life-threatening airway obstruction, especially in children. In a small number of cases, affected individuals are born with impaired drainage of the fluid that surrounds and protects the brain and spinal cord (the cerebrospinal fluid or CSF), resulting in a buildup of this fluid in the skull (occlusive hydrocephalus). It is unclear how this feature is related to the other signs and symptoms of congenital plasminogen deficiency.
Frequency
The prevalence of congenital plasminogen deficiency has been estimated at 1.6 per one million people. This condition is believed to be underdiagnosed, because growths in one area are often not recognized as being a feature of a disorder that affects many body systems. Mild cases likely never come to medical attention.
Causes
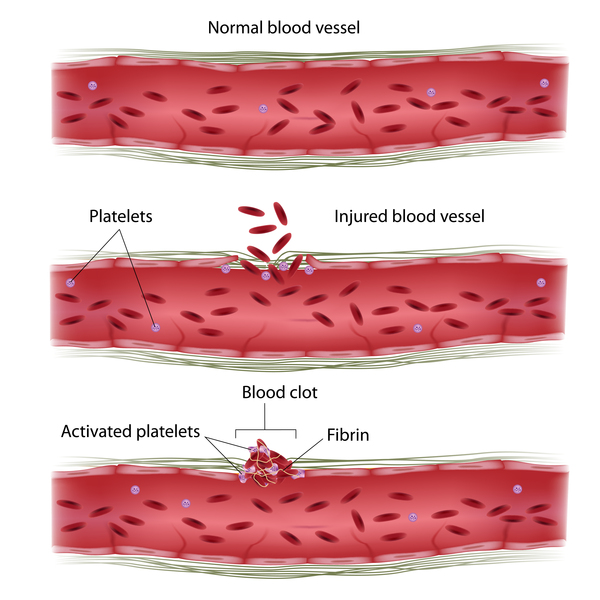
Congenital plasminogen deficiency is caused by mutations in the PLG gene. This gene provides instructions for making a protein called plasminogen. Enzymes called plasminogen activators convert plasminogen into the protein plasmin, which breaks down another protein called fibrin. Fibrin is the main protein involved in blood clots and is important for wound healing, creating the framework for normal tissue to grow back. Excess fibrin is broken down when no longer needed, and the new, more flexible normal tissue takes its place.
PLG gene mutations can decrease the amount of plasminogen that is produced, its function, or both. When the mutations affect plasminogen levels as well as the activity of the protein, affected individuals may be said to have type I congenital plasminogen deficiency, characterized by the ligneous growths previously described. People with mutations that result in normal levels of plasminogen with reduced activity are said to have type II congenital plasminogen deficiency or dysplasminogenemia. This form of the condition often has no symptoms.
A reduction in functional plasminogen results in less plasmin to break down fibrin, leading to a buildup of fibrin. The excess fibrin and the resulting inflammation of the tissue result in the inflamed woody growths characteristic of congenital plasminogen deficiency.
It is unclear why the excess fibrin builds up in the mucous membranes but does not usually result in abnormal clots in the blood vessels (thromboses). Researchers suggest that other enzymes in the blood may also break down fibrin, helping to compensate for the reduced plasminogen levels.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Hypoplasminogenemia
- Plasminogen deficiency, type I
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Klammt J, Kobelt L, Aktas D, Durak I, Gokbuget A, Hughes Q, Irkec M, Kurtulus I, Lapi E, Mechoulam H, Mendoza-Londono R, Palumbo JS, Steitzer H, Tabbara KF, Ozbek Z, Pucci N, Sotomayor T, Sturm M, Drogies T, Ziegler M, Schuster V. Identification of three novel plasminogen (PLG) gene mutations in a series of 23 patients with low PLG activity. Thromb Haemost. 2011 Mar;105(3):454-60. doi: 10.1160/TH10-04-0216. Epub 2010 Dec 21. Citation on PubMed
- Mehta R, Shapiro AD. Plasminogen deficiency. Haemophilia. 2008 Nov;14(6):1261-8. doi: 10.1111/j.1365-2516.2008.01825.x. Citation on PubMed
- Schuster V, Hugle B, Tefs K. Plasminogen deficiency. J Thromb Haemost. 2007 Dec;5(12):2315-22. doi: 10.1111/j.1538-7836.2007.02776.x. Epub 2007 Sep 26. Citation on PubMed
- Schuster V, Seregard S. Ligneous conjunctivitis. Surv Ophthalmol. 2003 Jul-Aug;48(4):369-88. doi: 10.1016/s0039-6257(03)00056-0. Citation on PubMed
- Tefs K, Gueorguieva M, Klammt J, Allen CM, Aktas D, Anlar FY, Aydogdu SD, Brown D, Ciftci E, Contarini P, Dempfle CE, Dostalek M, Eisert S, Gokbuget A, Gunhan O, Hidayat AA, Hugle B, Isikoglu M, Irkec M, Joss SK, Klebe S, Kneppo C, Kurtulus I, Mehta RP, Ornek K, Schneppenheim R, Seregard S, Sweeney E, Turtschi S, Veres G, Zeitler P, Ziegler M, Schuster V. Molecular and clinical spectrum of type I plasminogen deficiency: A series of 50 patients. Blood. 2006 Nov 1;108(9):3021-6. doi: 10.1182/blood-2006-04-017350. Epub 2006 Jul 18. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.