

Description
Congenital hyperinsulinism is a condition that causes individuals to have abnormally high levels of insulin. Insulin is a hormone that helps control levels of blood glucose, also called blood sugar. People with this condition have frequent episodes of low blood glucose (hypoglycemia). In infants and young children, these episodes are characterized by a lack of energy (lethargy), irritability, or difficulty feeding. Repeated episodes of low blood glucose increase the risk for serious complications such as breathing difficulties, seizures, intellectual disability, vision loss, brain damage, and coma.
The severity of congenital hyperinsulinism varies widely among affected individuals, even among members of the same family. About 60 percent of infants with this condition experience a hypoglycemic episode within the first month of life. Other affected children develop hypoglycemia by early childhood. Unlike typical episodes of hypoglycemia, which occur most often after periods without food (fasting) or after exercising, episodes of hypoglycemia in people with congenital hyperinsulinism can also occur after eating.
Frequency
Congenital hyperinsulinism affects approximately 1 in 50,000 newborns. This condition is more common in certain populations, affecting up to 1 in 2,500 newborns.
Causes
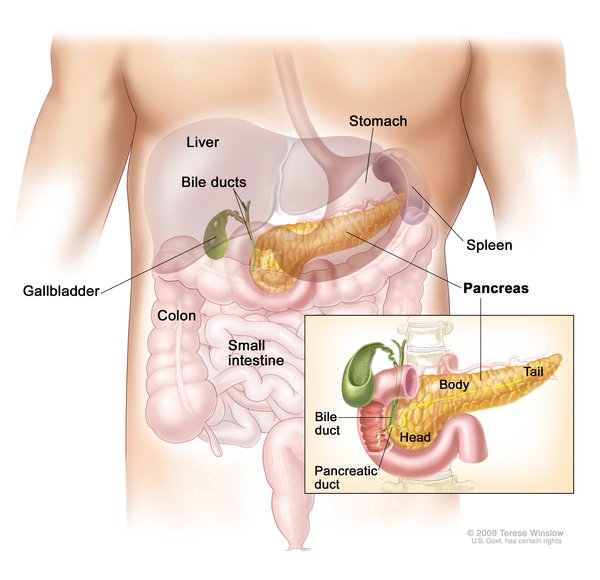
Congenital hyperinsulinism is caused by mutations in genes that regulate the release (secretion) of insulin, which is produced by beta cells in the pancreas. Insulin clears excess glucose from the bloodstream by passing glucose into cells to be used as energy.
Gene mutations that cause congenital hyperinsulinism lead to over-secretion of insulin from beta cells. Normally, insulin is secreted in response to the amount of glucose in the bloodstream: when glucose levels rise, so does insulin secretion. However, in people with congenital hyperinsulinism, insulin is secreted from beta cells regardless of the amount of glucose present in the blood. This excessive secretion of insulin results in glucose being rapidly removed from the bloodstream and passed into tissues such as muscle, liver, and fat. A lack of glucose in the blood results in frequent states of hypoglycemia in people with congenital hyperinsulinism. Insufficient blood glucose also deprives the brain of its primary source of fuel.
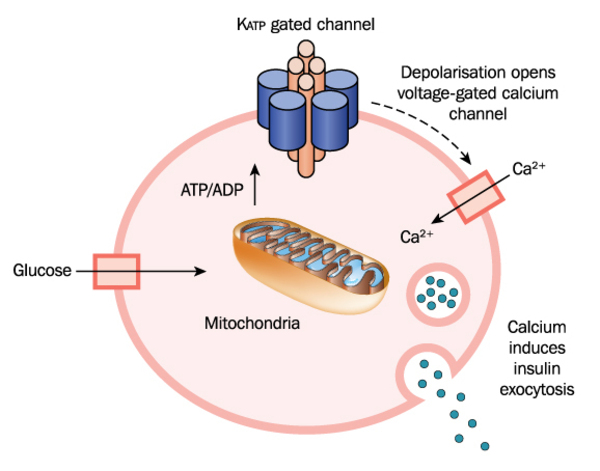
Mutations in at least nine genes have been found to cause congenital hyperinsulinism. Mutations in the ABCC8 gene are the most common known cause of the disorder. They account for this condition in approximately 40 percent of affected individuals. Less frequently, mutations in the KCNJ11 gene have been found in people with congenital hyperinsulinism. Mutations in each of the other genes associated with this condition account for only a small percentage of cases.
In approximately half of people with congenital hyperinsulinism, the cause is unknown.
Inheritance
Congenital hyperinsulinism can have different inheritance patterns, usually depending on the form of the condition. At least two forms of the condition have been identified. The most common form is the diffuse form, which occurs when all of the beta cells in the pancreas secrete too much insulin. The focal form of congenital hyperinsulinism occurs when only some of the beta cells over-secrete insulin.
Most often, the diffuse form of congenital hyperinsulinism is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Less frequently, the diffuse form is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.

The inheritance of the focal form of congenital hyperinsulinism is more complex. For most genes, both copies are turned on (active) in all cells, but for a small subset of genes, one of the two copies is turned off (inactive). Most people with the focal form of this condition inherit one copy of the mutated, inactive gene from their unaffected father. During embryonic development, a mutation occurs in the other, active copy of the gene. This second mutation is found within only some cells in the pancreas. As a result, some pancreatic beta cells have abnormal insulin secretion, while other beta cells function normally.
Other Names for This Condition
- Hyperinsulinemia hypoglycemia of infancy
- Infancy hyperinsulinemia hypoglycemia
- Neonatal hyperinsulinism
- Persistent hyperinsulinemia hypoglycemia of infancy
- Persistent hyperinsulinemic hypoglycemia
- PHHI hypoglycemia
Additional Information & Resources
Genetic Testing Information
- Genetic Testing Registry: Exercise-induced hyperinsulinism

- Genetic Testing Registry: Familial hyperinsulinism
- Genetic Testing Registry: Hyperinsulinemic hypoglycemia, familial, 2
- Genetic Testing Registry: Hyperinsulinemic hypoglycemia, familial, 3
- Genetic Testing Registry: Hyperinsulinism due to INSR deficiency
- Genetic Testing Registry: Hyperinsulinemic hypoglycemia, familial, 1
- Genetic Testing Registry: Hyperinsulinemic hypoglycemia, familial, 4
- Genetic Testing Registry: Hyperinsulinism-hyperammonemia syndrome
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
- HYPERINSULINEMIC HYPOGLYCEMIA, FAMILIAL, 1; HHF1
- HYPERINSULINEMIC HYPOGLYCEMIA, FAMILIAL, 2; HHF2
- HYPERINSULINEMIC HYPOGLYCEMIA, FAMILIAL, 3; HHF3
- HYPERINSULINEMIC HYPOGLYCEMIA, FAMILIAL, 4; HHF4
- HYPERINSULINEMIC HYPOGLYCEMIA, FAMILIAL, 5; HHF5
- HYPERINSULINEMIC HYPOGLYCEMIA, FAMILIAL, 6; HHF6
- HYPERINSULINEMIC HYPOGLYCEMIA, FAMILIAL, 7; HHF7
Scientific Articles on PubMed
References
- Huopio H, Shyng SL, Otonkoski T, Nichols CG. K(ATP) channels and insulin secretion disorders. Am J Physiol Endocrinol Metab. 2002 Aug;283(2):E207-16. doi: 10.1152/ajpendo.00047.2002. Citation on PubMed
- James C, Kapoor RR, Ismail D, Hussain K. The genetic basis of congenital hyperinsulinism. J Med Genet. 2009 May;46(5):289-99. doi: 10.1136/jmg.2008.064337. Epub 2009 Mar 1. Citation on PubMed
- Kapoor RR, James C, Hussain K. Advances in the diagnosis and management of hyperinsulinemic hypoglycemia. Nat Clin Pract Endocrinol Metab. 2009 Feb;5(2):101-12. doi: 10.1038/ncpendmet1046. Citation on PubMed
- Meissner T, Wendel U, Burgard P, Schaetzle S, Mayatepek E. Long-term follow-up of 114 patients with congenital hyperinsulinism. Eur J Endocrinol. 2003 Jul;149(1):43-51. doi: 10.1530/eje.0.1490043. Citation on PubMed
- Mohamed Z, Arya VB, Hussain K. Hyperinsulinaemic hypoglycaemia:genetic mechanisms, diagnosis and management. J Clin Res Pediatr Endocrinol. 2012 Dec;4(4):169-81. doi: 10.4274/jcrpe.821. Epub 2012 Oct 2. Citation on PubMed or Free article on PubMed Central
- Pinney SE, MacMullen C, Becker S, Lin YW, Hanna C, Thornton P, Ganguly A, Shyng SL, Stanley CA. Clinical characteristics and biochemical mechanisms of congenital hyperinsulinism associated with dominant KATP channel mutations. J Clin Invest. 2008 Aug;118(8):2877-86. doi: 10.1172/JCI35414. Citation on PubMed or Free article on PubMed Central
- Sandal T, Laborie LB, Brusgaard K, Eide SA, Christesen HB, Sovik O, Njolstad PR, Molven A. The spectrum of ABCC8 mutations in Norwegian patients with congenital hyperinsulinism of infancy. Clin Genet. 2009 May;75(5):440-8. doi: 10.1111/j.1399-0004.2009.01152.x. Citation on PubMed
- Snider KE, Becker S, Boyajian L, Shyng SL, MacMullen C, Hughes N, Ganapathy K, Bhatti T, Stanley CA, Ganguly A. Genotype and phenotype correlations in 417 children with congenital hyperinsulinism. J Clin Endocrinol Metab. 2013 Feb;98(2):E355-63. doi: 10.1210/jc.2012-2169. Epub 2012 Dec 28. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.