

Description
CLPB deficiency is a rare disorder characterized by neurological problems and a shortage of infection-fighting white blood cells (neutropenia). Signs and symptoms of the condition develop by early childhood, and their severity varies widely among affected individuals.
In the most severely affected individuals, features of CLPB deficiency are apparent in infancy and sometimes at birth. Affected babies have serious neurological problems, which can include an exaggerated startle reaction (hyperekplexia) to unexpected stimuli such as loud noises, reduced movement, muscle tone that is either decreased (hypotonia) or increased (hypertonia), swallowing problems, difficulty breathing, and recurrent seizures (epilepsy). These babies may also have movement abnormalities, such as difficulty coordinating movements (ataxia), involuntary tensing of the muscles (dystonia), or uncontrolled movements of the body (dyskinesia). In addition, these babies have recurrent, life-threatening infections due to severe neutropenia. Affected individuals are at risk of developing a blood cell disorder called myelodysplastic syndrome or a form of blood cancer called leukemia. Because of their severe health problems, affected infants usually live only a few weeks or months.
Moderately affected individuals have neurological problems similar to those described above, although they are less severe. They include hypotonia, muscle stiffness (spasticity), and movement abnormalities. Other features of moderate CLPB deficiency include epilepsy and mild to severe intellectual disability. Neutropenia in these individuals can lead to recurrent infections, although they are not life-threatening.
Mildly affected individuals have no neurological problems, and although they have neutropenia, it does not increase the risk of infections. Some people with mild CLPB deficiency develop deposits of calcium in the kidneys (nephrocalcinosis) or kidney (renal) cysts.
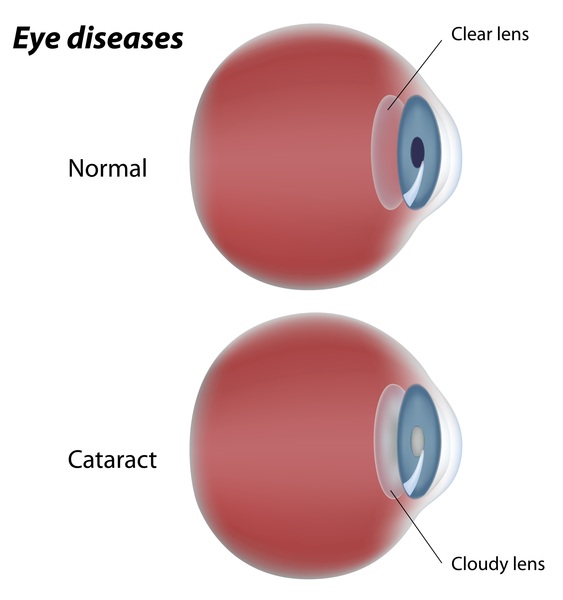
Many people with mild, moderate, or severe CLPB deficiency have clouding of the lenses of the eyes (cataracts) from birth (congenital) or beginning in infancy.
CLPB deficiency is associated with increased levels of a substance called 3-methylglutaconic acid in the urine (3-methylglutaconic aciduria). This abnormality, which provides a clue to the diagnosis, does not appear to cause any health problems.
Frequency
CLPB deficiency is a rare disorder; the prevalence is not known. At least 26 cases have been reported in the medical literature.
Causes
CLPB deficiency is caused by mutations in the CLPB gene, which provides instructions for making a protein whose function is unknown. Based on its similarity to a protein in other organisms, the CLPB protein is thought to help unfold misfolded proteins so they can be refolded correctly. If not fixed, misfolded proteins cannot function properly and may be damaging to cells.
CLPB gene mutations likely reduce or eliminate the amount of functional CLPB protein. The severity of the condition is thought to be related to the amount of functional protein remaining: severe CLPB deficiency is likely caused by a complete absence of CLPB protein, while moderate and mild CLPB deficiency result when some functional CLPB protein is produced. Researchers are unsure how reduction or absence of this protein leads to the signs and symptoms of CLPB deficiency.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- 3-methylglutaconic aciduria type 7
- 3-methylglutaconic aciduria type VII
- 3-methylglutaconic aciduria with cataracts, neurologic involvement and neutropenia
- 3-methylglutaconic aciduria-cataract-neurologic involvement-neutropenia syndrome
- MEGCANN
- MGA7
- MGCA7
Additional Information & Resources
Genetic Testing Information
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Capo-Chichi JM, Boissel S, Brustein E, Pickles S, Fallet-Bianco C, Nassif C, Patry L, Dobrzeniecka S, Liao M, Labuda D, Samuels ME, Hamdan FF, Vande Velde C, Rouleau GA, Drapeau P, Michaud JL. Disruption of CLPB is associated with congenital microcephaly, severe encephalopathy and 3-methylglutaconic aciduria. J Med Genet. 2015 May;52(5):303-11. doi: 10.1136/jmedgenet-2014-102952. Epub 2015 Feb 3. Citation on PubMed
- Kanabus M, Shahni R, Saldanha JW, Murphy E, Plagnol V, Hoff WV, Heales S, Rahman S. Bi-allelic CLPB mutations cause cataract, renal cysts, nephrocalcinosis and 3-methylglutaconic aciduria, a novel disorder of mitochondrial protein disaggregation. J Inherit Metab Dis. 2015 Mar;38(2):211-9. doi: 10.1007/s10545-015-9813-0. Epub 2015 Jan 18. Citation on PubMed
- Saunders C, Smith L, Wibrand F, Ravn K, Bross P, Thiffault I, Christensen M, Atherton A, Farrow E, Miller N, Kingsmore SF, Ostergaard E. CLPB variants associated with autosomal-recessive mitochondrial disorder with cataract, neutropenia, epilepsy, and methylglutaconic aciduria. Am J Hum Genet. 2015 Feb 5;96(2):258-65. doi: 10.1016/j.ajhg.2014.12.020. Epub 2015 Jan 15. Citation on PubMed or Free article on PubMed Central
- Wortmann SB, Wevers RA. CLPB Deficiency. 2016 Nov 22 [updated 2022 Mar 10]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK396257/ Citation on PubMed
- Wortmann SB, Zietkiewicz S, Kousi M, Szklarczyk R, Haack TB, Gersting SW, Muntau AC, Rakovic A, Renkema GH, Rodenburg RJ, Strom TM, Meitinger T, Rubio-Gozalbo ME, Chrusciel E, Distelmaier F, Golzio C, Jansen JH, van Karnebeek C, Lillquist Y, Lucke T, Ounap K, Zordania R, Yaplito-Lee J, van Bokhoven H, Spelbrink JN, Vaz FM, Pras-Raves M, Ploski R, Pronicka E, Klein C, Willemsen MA, de Brouwer AP, Prokisch H, Katsanis N, Wevers RA. CLPB mutations cause 3-methylglutaconic aciduria, progressive brain atrophy, intellectual disability, congenital neutropenia, cataracts, movement disorder. Am J Hum Genet. 2015 Feb 5;96(2):245-57. doi: 10.1016/j.ajhg.2014.12.013. Epub 2015 Jan 15. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.