

Description
CLN10 disease is a severe disorder that primarily affects the nervous system. Individuals with this condition typically show signs and symptoms soon after birth. These signs and symptoms can include muscle rigidity, respiratory failure, and prolonged episodes of seizure activity that last several minutes (status epilepticus). It is likely that some affected individuals also have seizures before birth while in the womb. Infants with CLN10 disease have unusually small heads (microcephaly) with brains that may be less than half the normal size. There is a loss of brain cells in areas that coordinate movement (the cerebellum) and control thinking and emotions (the cerebral cortex). Nerve cells in the brain also lack a fatty substance called myelin, which protects them and promotes efficient transmission of nerve impulses. Infants with CLN10 disease often die hours to weeks after birth.
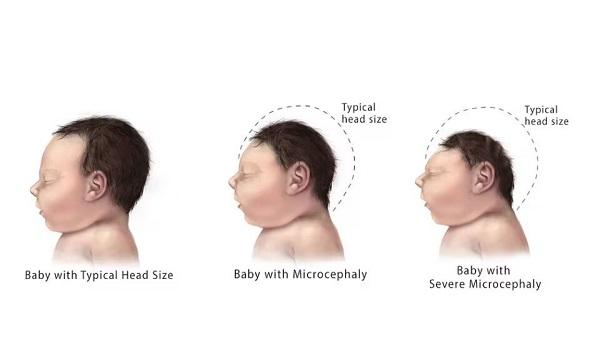

In some individuals with CLN10 disease, the condition does not appear until later in life, between late infancy and adulthood. These individuals have a gradual loss of brain cells and often develop problems with balance and coordination (ataxia), loss of speech, a progressive loss in intellectual functioning (cognitive decline), and vision loss. Individuals with later-onset CLN10 disease have a shortened lifespan, depending on when their signs and symptoms first started.
CLN10 disease is one of a group of disorders known as neuronal ceroid lipofuscinoses (NCLs). All of these disorders affect the nervous system and typically cause progressive problems with vision, movement, and thinking ability. The different NCLs are distinguished by their genetic cause. Each disease type is given the designation "CLN," meaning ceroid lipofuscinosis, neuronal, and then a number to indicate its subtype.
Frequency
The prevalence of CLN10 disease is unknown; at least 11 cases have been described.
Causes
Mutations in the CTSD gene cause CLN10 disease. The CTSD gene provides instructions for making an enzyme called cathepsin D. Cathepsin D is one of a family of cathepsin proteins that act as protease enzymes, which modify proteins by cutting them apart. Cathepsin D is found in many types of cells and is active in lysosomes, which are compartments within cells that digest and recycle different types of molecules. By cutting proteins apart, cathepsin D can break down certain proteins, turn on (activate) other proteins, and regulate self-destruction of the cell (apoptosis).

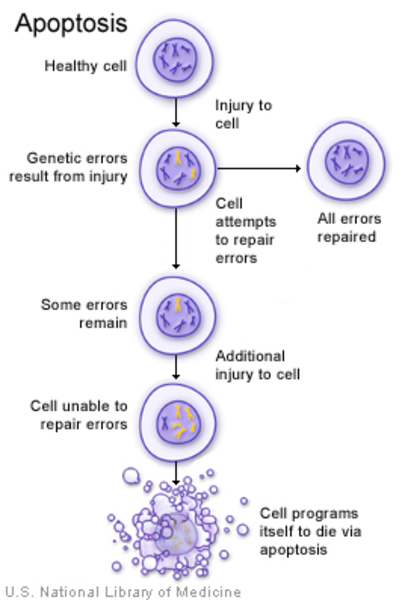
CTSD gene mutations found to cause CLN10 disease that is present at birth lead to a complete lack of cathepsin D enzyme activity. As a result, proteins and fats are not broken down properly and abnormally accumulate within lysosomes. While accumulations of these substances occurs in cells throughout the body, nerve cells appear to be particularly vulnerable to damage caused by the abnormal cell materials. Early and widespread loss of nerve cells in CLN10 disease leads to severe signs and symptoms and death in infancy.
In the later-onset cases of CLN10 disease, CTSD gene mutations likely result in the production of a cathepsin D enzyme whose function is greatly reduced but not eliminated. As a result, some proteins and fats are broken down by the enzyme, so it takes longer for these substances to accumulate in lysosomes and cause nerve cell death.

Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Cathepsin D deficiency
- Cathepsin D deficient neuronal ceroid lipofuscinosis
- CLN10
- Congenital neuronal ceroid lipofuscinosis
- Neuronal ceroid lipofuscinosis 10
- Neuronal ceroid lipofuscinosis due to cathepsin D deficiency
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Anderson GW, Goebel HH, Simonati A. Human pathology in NCL. Biochim Biophys Acta. 2013 Nov;1832(11):1807-26. doi: 10.1016/j.bbadis.2012.11.014. Epub 2012 Nov 29. Citation on PubMed
- Jalanko A, Braulke T. Neuronal ceroid lipofuscinoses. Biochim Biophys Acta. 2009 Apr;1793(4):697-709. doi: 10.1016/j.bbamcr.2008.11.004. Epub 2008 Nov 24. Citation on PubMed
- Kousi M, Lehesjoki AE, Mole SE. Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum Mutat. 2012 Jan;33(1):42-63. doi: 10.1002/humu.21624. Epub 2011 Nov 16. Citation on PubMed
- Siintola E, Partanen S, Stromme P, Haapanen A, Haltia M, Maehlen J, Lehesjoki AE, Tyynela J. Cathepsin D deficiency underlies congenital human neuronal ceroid-lipofuscinosis. Brain. 2006 Jun;129(Pt 6):1438-45. doi: 10.1093/brain/awl107. Epub 2006 May 2. Citation on PubMed
- Steinfeld R, Reinhardt K, Schreiber K, Hillebrand M, Kraetzner R, Bruck W, Saftig P, Gartner J. Cathepsin D deficiency is associated with a human neurodegenerative disorder. Am J Hum Genet. 2006 Jun;78(6):988-98. doi: 10.1086/504159. Epub 2006 Mar 29. Citation on PubMed or Free article on PubMed Central
- Williams RE, Mole SE. New nomenclature and classification scheme for the neuronal ceroid lipofuscinoses. Neurology. 2012 Jul 10;79(2):183-91. doi: 10.1212/WNL.0b013e31825f0547. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.