

Description
Childhood absence epilepsy is a condition characterized by recurrent seizures (epilepsy). This condition begins in childhood, usually between ages 3 and 8. Affected children have absence seizures (also known as petit mal seizures), which are brief episodes of impaired consciousness that look like staring spells. During seizures, children are not aware of and do not respond to people or activities around them. The seizures usually last several seconds and they occur often, up to 200 times each day.
Some affected individuals have febrile seizures before they develop childhood absence epilepsy. Febrile seizures are involuntary muscle contractions (convulsions) brought on by a high body temperature (fever).
In most people with childhood absence epilepsy, the absence seizures disappear in adolescence. However, some affected individuals continue to have absence seizures into adulthood, or they may develop generalized tonic-clonic seizures, which cause muscle rigidity, convulsions, and loss of consciousness, or myoclonic seizures, which are characterized by rapid, uncontrolled muscle jerks.
Frequency
Childhood absence epilepsy affects 2 to 8 in 100,000 children under age 15 each year. The condition is more common in girls than in boys.
Causes
The genetics of childhood absence epilepsy are complex and not completely understood. It is thought that multiple genetic changes or a combination of genetic and environmental factors contribute to development of the condition. Most genetic changes associated with childhood absence epilepsy are rare, having been found in only a small number of affected individuals. Each genetic change appears to play a role in some populations but not others.
Several genes associated with childhood absence epilepsy provide instructions for making pieces (subunits) of the GABAA receptor protein. The GABAA receptor acts as a channel that allows negatively charged chlorine atoms (chloride ions) to cross the cell membrane. The influx of chloride ions in nerve cells (neurons) in developed brains creates an environment that blocks (inhibits) signaling between neurons and prevents the brain from being overloaded with too many signals. Mutations in GABAA receptor subunit genes lead to production of altered subunit proteins that cannot form functional receptors, so fewer GABAA receptors are available. As a result, neurons become overloaded with signals. Researchers believe that the overstimulation of certain neurons in the brain triggers the abnormal brain activity associated with seizures.


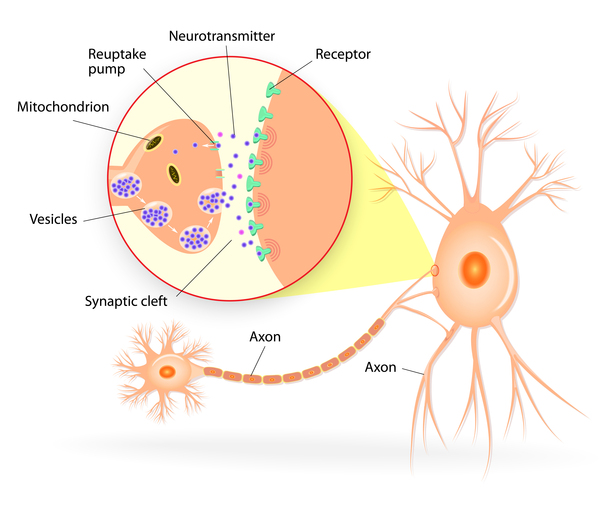
Problems with another type of ion channel, called a calcium channel, are also associated with childhood absence epilepsy. Calcium channels transport positively charged calcium atoms (calcium ions) into cells. These channels help control the release of neurotransmitters, which are chemicals that relay signals from one neuron to another. Mutations that result in overactive calcium channels cause certain neurons to become overstimulated, triggering seizures.
Mutations in other genes that do not provide instructions for making ion channels have also been associated with childhood absence epilepsy. It is not clear how these changes are involved in the development of absence seizures.
Inheritance
Because childhood absence epilepsy appears to be a complex disease without a single genetic cause, it does not have a straightforward pattern of inheritance. When associated with mutations in GABAA receptor or calcium channel genes, it seems to follow an autosomal dominant inheritance pattern, which means one copy of the altered gene in each cell is sufficient to increase the likelihood of the disorder. Some people who have the altered gene never develop the condition, a situation known as reduced penetrance.
Other Names for This Condition
- Absence epilepsy, childhood
- Petit mal epilepsy
- Pykno-epilepsy
- Pyknolepsy
Additional Information & Resources
Genetic Testing Information
- Genetic Testing Registry: Epilepsy, childhood absence 4

- Genetic Testing Registry: Epilepsy, childhood absence, JRK related
- Genetic Testing Registry: Epilepsy, childhood absence, susceptibility to, 1
- Genetic Testing Registry: EPILEPSY, CHILDHOOD ABSENCE, SUSCEPTIBILITY TO, 2
- Genetic Testing Registry: Epilepsy, childhood absence, susceptibility to, 5
- Genetic Testing Registry: Epilepsy, childhood absence, susceptibility to, 6
Patient Support and Advocacy Resources
Clinical Trials
Scientific Articles on PubMed
References
- Crunelli V, Leresche N. Childhood absence epilepsy: genes, channels, neurons and networks. Nat Rev Neurosci. 2002 May;3(5):371-82. doi: 10.1038/nrn811. Citation on PubMed
- Hirose S. Mutant GABA(A) receptor subunits in genetic (idiopathic) epilepsy. Prog Brain Res. 2014;213:55-85. doi: 10.1016/B978-0-444-63326-2.00003-X. Citation on PubMed
- Kang JQ, Macdonald RL. Molecular Pathogenic Basis for GABRG2 Mutations Associated With a Spectrum of Epilepsy Syndromes, From Generalized Absence Epilepsy to Dravet Syndrome. JAMA Neurol. 2016 Aug 1;73(8):1009-16. doi: 10.1001/jamaneurol.2016.0449. Citation on PubMed or Free article on PubMed Central
- Yalcin O. Genes and molecular mechanisms involved in the epileptogenesis of idiopathic absence epilepsies. Seizure. 2012 Mar;21(2):79-86. doi: 10.1016/j.seizure.2011.12.002. Epub 2011 Dec 27. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.