

Description
Canavan disease is a rare inherited disorder that damages the ability of nerve cells (neurons) in the brain to send and receive messages. This disease is one of a group of genetic disorders called leukodystrophies. Leukodystrophies disrupt the growth or maintenance of the myelin sheath, which is the covering that protects nerves and promotes the efficient transmission of nerve impulses.

Neonatal/infantile Canavan disease is the most common and most severe form of the condition. Affected infants appear normal for the first few months of life, but by age 3 to 5 months, problems with development become noticeable. These infants usually do not develop motor skills such as turning over, controlling head movement, and sitting without support. Other common features of this condition include weak muscle tone (hypotonia), an unusually large head size (macrocephaly), and irritability. Feeding and swallowing difficulties, seizures, and sleep disturbances may also develop.
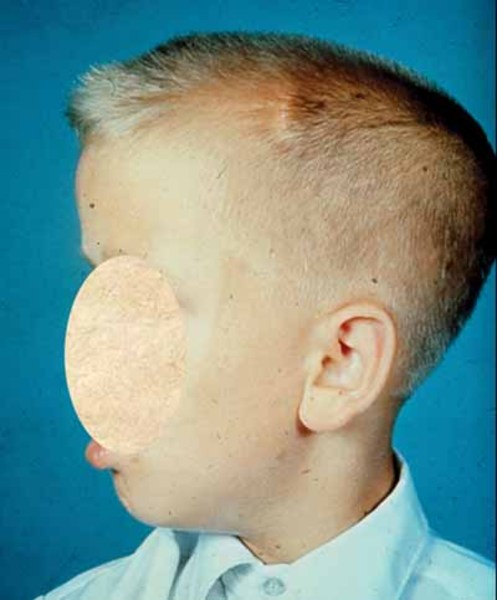
The mild/juvenile form of Canavan disease is less common. Affected individuals have mildly delayed development of speech and motor skills starting in childhood. These delays may be so mild and nonspecific that they are never recognized as being caused by Canavan disease.
The life expectancy for people with Canavan disease varies. Most people with the neonatal/infantile form live only into childhood, although some survive into adolescence or beyond. People with the mild/juvenile form do not appear to have a shortened lifespan.
Frequency
While this condition occurs in people of all ethnic backgrounds, it is most common in people of Ashkenazi (eastern and central European) Jewish heritage. Studies suggest that this disorder affects 1 in 6,400 to 13,500 people in the Ashkenazi Jewish population. The incidence in other populations is unknown.
Causes
Mutations in the ASPA gene cause Canavan disease. The ASPA gene provides instructions for making an enzyme called aspartoacylase. This enzyme normally breaks down a compound called N-acetyl-L-aspartic acid (NAA), which is predominantly found in neurons in the brain. The function of NAA is unclear. Researchers had suspected that it played a role in the production of the myelin sheath, but recent studies suggest that NAA does not have this function. The enzyme may instead be involved in the transport of water molecules out of neurons.
Mutations in the ASPA gene reduce the function of aspartoacylase, which prevents the normal breakdown of NAA. The mutations that cause the neonatal/infantile form of Canavan disease severely impair the enzyme's activity, allowing NAA to build up to high levels in the brain. The mutations that cause the mild/juvenile form of the disorder have milder effects on the enzyme's activity, leading to less accumulation of NAA.
An excess of NAA in the brain is associated with the signs and symptoms of Canavan disease. Studies suggest that if NAA is not broken down properly, the resulting chemical imbalance interferes with the formation of the myelin sheath as the nervous system develops. A buildup of NAA also leads to the progressive destruction of existing myelin sheaths. Nerves without this protective covering malfunction, which disrupts normal brain development.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- ACY2 deficiency
- Aminoacylase 2 deficiency
- Aspa deficiency
- Aspartoacylase deficiency
- Canavan's disease
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Baslow MH, Guilfoyle DN. Canavan disease, a rare early-onset human spongiform leukodystrophy: insights into its genesis and possible clinical interventions. Biochimie. 2013 Apr;95(4):946-56. doi: 10.1016/j.biochi.2012.10.023. Epub 2012 Nov 11. Citation on PubMed
- Feigenbaum A, Moore R, Clarke J, Hewson S, Chitayat D, Ray PN, Stockley TL. Canavan disease: carrier-frequency determination in the Ashkenazi Jewish population and development of a novel molecular diagnostic assay. Am J Med Genet A. 2004 Jan 15;124A(2):142-7. doi: 10.1002/ajmg.a.20334. Citation on PubMed
- Guo F, Bannerman P, Mills Ko E, Miers L, Xu J, Burns T, Li S, Freeman E, McDonough JA, Pleasure D. Ablating N-acetylaspartate prevents leukodystrophy in a Canavan disease model. Ann Neurol. 2015 May;77(5):884-8. doi: 10.1002/ana.24392. Epub 2015 Mar 27. Citation on PubMed
- Janson CG, McPhee SW, Francis J, Shera D, Assadi M, Freese A, Hurh P, Haselgrove J, Wang DJ, Bilaniuk L, Leone P. Natural history of Canavan disease revealed by proton magnetic resonance spectroscopy (1H-MRS) and diffusion-weighted MRI. Neuropediatrics. 2006 Aug;37(4):209-21. doi: 10.1055/s-2006-924734. Citation on PubMed
- Madhavarao CN, Arun P, Moffett JR, Szucs S, Surendran S, Matalon R, Garbern J, Hristova D, Johnson A, Jiang W, Namboodiri MA. Defective N-acetylaspartate catabolism reduces brain acetate levels and myelin lipid synthesis in Canavan's disease. Proc Natl Acad Sci U S A. 2005 Apr 5;102(14):5221-6. doi: 10.1073/pnas.0409184102. Epub 2005 Mar 22. Citation on PubMed or Free article on PubMed Central
- Nagy A, Bley AE, Eichler F. Canavan Disease. 1999 Sep 16 [updated 2023 Dec 21]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1234/ Citation on PubMed
- Namboodiri AM, Peethambaran A, Mathew R, Sambhu PA, Hershfield J, Moffett JR, Madhavarao CN. Canavan disease and the role of N-acetylaspartate in myelin synthesis. Mol Cell Endocrinol. 2006 Jun 27;252(1-2):216-23. doi: 10.1016/j.mce.2006.03.016. Epub 2006 May 2. Citation on PubMed
- Surendran S, Bamforth FJ, Chan A, Tyring SK, Goodman SI, Matalon R. Mild elevation of N-acetylaspartic acid and macrocephaly: diagnostic problem. J Child Neurol. 2003 Nov;18(11):809-12. doi: 10.1177/08830738030180111601. Citation on PubMed
- Surendran S, Michals-Matalon K, Quast MJ, Tyring SK, Wei J, Ezell EL, Matalon R. Canavan disease: a monogenic trait with complex genomic interaction. Mol Genet Metab. 2003 Sep-Oct;80(1-2):74-80. doi: 10.1016/j.ymgme.2003.08.015. Citation on PubMed
- Tacke U, Olbrich H, Sass JO, Fekete A, Horvath J, Ziyeh S, Kleijer WJ, Rolland MO, Fisher S, Payne S, Vargiami E, Zafeiriou DI, Omran H. Possible genotype-phenotype correlations in children with mild clinical course of Canavan disease. Neuropediatrics. 2005 Aug;36(4):252-5. doi: 10.1055/s-2005-865865. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.