

Description
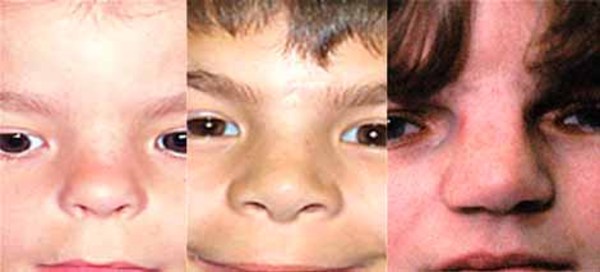
Axenfeld-Rieger syndrome is primarily an eye disorder, although it can also affect other parts of the body. This condition is characterized by abnormalities of the front part of the eye, an area known as the anterior segment. For example, the colored part of the eye (the iris), may be thin or poorly developed. The iris normally has a single central hole, called the pupil, through which light enters the eye. People with Axenfeld-Rieger syndrome often have a pupil that is off-center (corectopia) or extra holes in the iris that can look like multiple pupils (polycoria). This condition can also cause abnormalities of the cornea, which is the clear front covering of the eye.
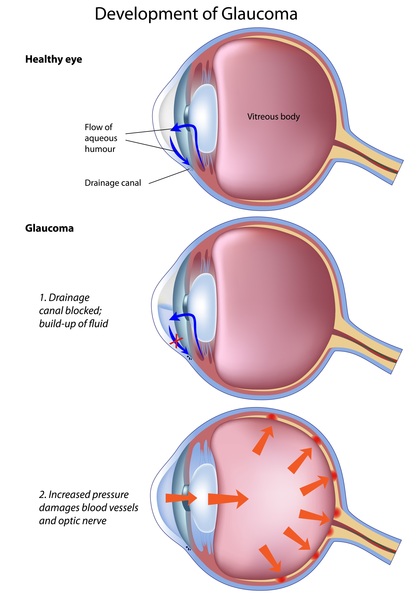
About half of affected individuals develop glaucoma, a serious condition that increases pressure inside the eye. When glaucoma occurs with Axenfeld-Rieger syndrome, it most often develops in late childhood or adolescence, although it can occur as early as infancy. Glaucoma can cause vision loss or blindness.
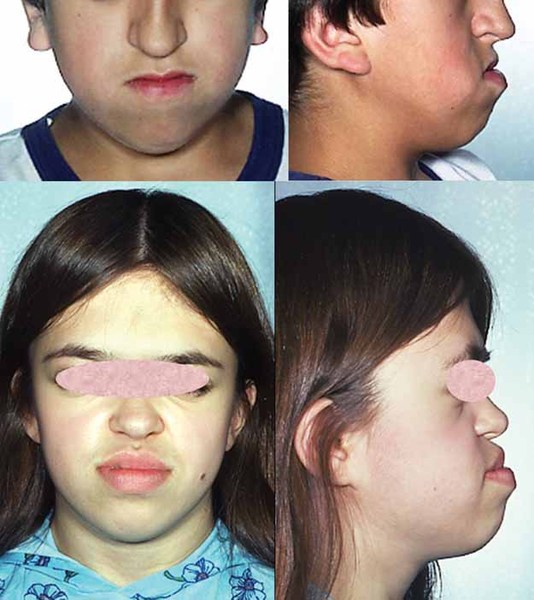
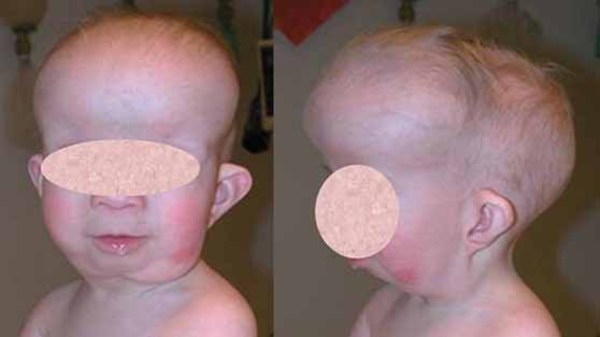
The signs and symptoms of Axenfeld-Rieger syndrome can also affect other parts of the body. Many affected individuals have distinctive facial features such as widely spaced eyes (hypertelorism); a flattened mid-face with a broad, flat nasal bridge; and a prominent forehead. The condition is also associated with dental abnormalities including unusually small teeth (microdontia) or fewer than normal teeth (oligodontia). Some people with Axenfeld-Rieger syndrome have extra folds of skin around their belly button (redundant periumbilical skin). Other, less common features can include heart defects, the opening of the urethra on the underside of the penis (hypospadias), narrowing of the anus (anal stenosis), and abnormalities of the pituitary gland that can result in slow growth.


Researchers have described at least three types of Axenfeld-Rieger syndrome. The types, which are numbered 1 through 3, are distinguished by their genetic cause.
Frequency
Axenfeld-Rieger syndrome has an estimated prevalence of 1 in 200,000 people.
Causes
Axenfeld-Rieger syndrome results from mutations in at least two known genes, PITX2 and FOXC1. PITX2 gene mutations cause type 1, and FOXC1 gene mutations cause type 3. The gene associated with type 2 is likely located on chromosome 13, but it has not been identified. The proteins produced from the PITX2 and FOXC1 genes are transcription factors, which means they attach (bind) to DNA and help control the activity of other genes. These transcription factors are active before birth in the developing eye and in other parts of the body. They appear to play important roles in embryonic development, particularly in the formation of structures in the anterior segment of the eye.
Mutations in either the PITX2 or FOXC1 gene disrupt the activity of other genes that are needed for normal development. Impaired regulation of these genes leads to problems in the formation of the anterior segment of the eye and other parts of the body. These developmental abnormalities underlie the characteristic features of Axenfeld-Rieger syndrome. Affected individuals with PITX2 gene mutations are more likely than those with FOXC1 gene mutations to have abnormalities affecting parts of the body other than the eye.
Some people with Axenfeld-Rieger syndrome do not have identified mutations in the PITX2 or FOXC1 genes. In these individuals, the cause of the condition is unknown. Other as-yet-unidentified genes may also cause Axenfeld-Rieger syndrome.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.

Other Names for This Condition
- ARS
- Axenfeld and Rieger anomaly
- Axenfeld anomaly
- Axenfeld syndrome
- AXRA
- AXRS
- Rieger anomaly
- Rieger syndrome
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Chang TC, Summers CG, Schimmenti LA, Grajewski AL. Axenfeld-Rieger syndrome: new perspectives. Br J Ophthalmol. 2012 Mar;96(3):318-22. doi: 10.1136/bjophthalmol-2011-300801. Epub 2011 Dec 23. Citation on PubMed
- D'haene B, Meire F, Claerhout I, Kroes HY, Plomp A, Arens YH, de Ravel T, Casteels I, De Jaegere S, Hooghe S, Wuyts W, van den Ende J, Roulez F, Veenstra-Knol HE, Oldenburg RA, Giltay J, Verheij JB, de Faber JT, Menten B, De Paepe A, Kestelyn P, Leroy BP, De Baere E. Expanding the spectrum of FOXC1 and PITX2 mutations and copy number changes in patients with anterior segment malformations. Invest Ophthalmol Vis Sci. 2011 Jan 21;52(1):324-33. doi: 10.1167/iovs.10-5309. Citation on PubMed
- Hjalt TA, Semina EV. Current molecular understanding of Axenfeld-Rieger syndrome. Expert Rev Mol Med. 2005 Nov 8;7(25):1-17. doi: 10.1017/S1462399405010082. Citation on PubMed
- Idrees F, Vaideanu D, Fraser SG, Sowden JC, Khaw PT. A review of anterior segment dysgeneses. Surv Ophthalmol. 2006 May-Jun;51(3):213-31. doi: 10.1016/j.survophthal.2006.02.006. Citation on PubMed
- Phillips JC, del Bono EA, Haines JL, Pralea AM, Cohen JS, Greff LJ, Wiggs JL. A second locus for Rieger syndrome maps to chromosome 13q14. Am J Hum Genet. 1996 Sep;59(3):613-9. Citation on PubMed or Free article on PubMed Central
- Strungaru MH, Dinu I, Walter MA. Genotype-phenotype correlations in Axenfeld-Rieger malformation and glaucoma patients with FOXC1 and PITX2 mutations. Invest Ophthalmol Vis Sci. 2007 Jan;48(1):228-37. doi: 10.1167/iovs.06-0472. Citation on PubMed
- Tumer Z, Bach-Holm D. Axenfeld-Rieger syndrome and spectrum of PITX2 and FOXC1 mutations. Eur J Hum Genet. 2009 Dec;17(12):1527-39. doi: 10.1038/ejhg.2009.93. Epub 2009 Jun 10. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.