

Description
Au-Kline syndrome is a condition that affects many body systems. Individuals with this condition typically have weak muscle tone (hypotonia), intellectual disability, and delayed development. Speech is delayed in children with Au-Kline syndrome, and some are able to say only one or a few words or are never able to speak. In addition, affected children learn to walk later than usual, and some are never able to walk on their own.
Individuals with Au-Kline syndrome can have distinctive facial features, including long openings of the eyelids (long palpebral fissures), drooping eyelids (ptosis), and shallow eye sockets. Other common facial features in this condition include a broad nasal bridge, a mouth with the outer corners turned downward and often held in an open position, and a deep groove down the middle of the tongue. Less common abnormalities include premature joining of certain skull bones (craniosynostosis) in affected infants, an opening or unusually high arch in the roof of the mouth (cleft or high-arched palate), a split in the soft flap of tissue that hangs from the back of the mouth (bifid uvula), and missing teeth (oligodontia).

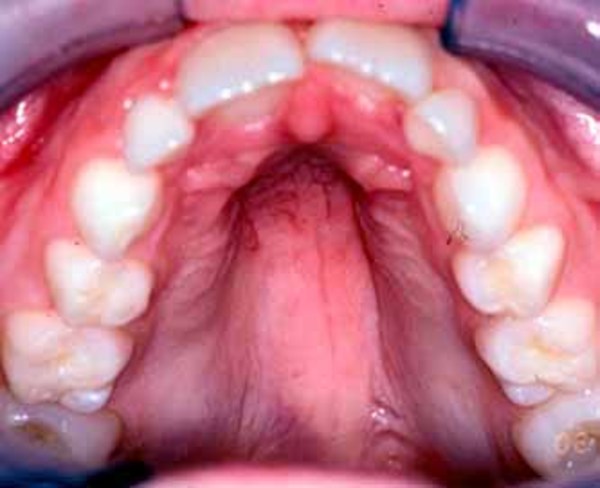
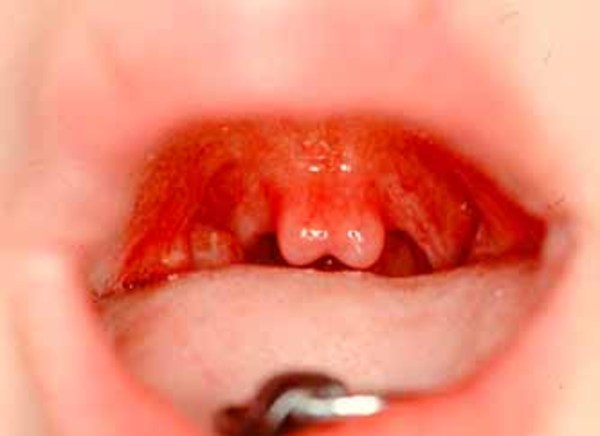
Malformations of the heart, blood vessels, kidneys, or bones can also occur in people with Au-Kline syndrome. For example, in some affected individuals, the large blood vessel that distributes blood from the heart to the rest of the body (the aorta) becomes weakened and stretched (aortic dilatation), which can be life-threatening. Some people with Au-Kline syndrome have an abnormal curvature of the spine (scoliosis). In addition, affected individuals may have difficulty feeding or poor vision.
Au-Kline syndrome can sometimes affect the autonomic nervous system, which controls involuntary body functions, such as digestion and regulation of body temperature. In people with Au-Kline syndrome, abnormalities in this system can lead to digestive problems, difficulty feeling pain, abnormal sweating, and an inability to adjust to high heat in people with Au-Kline syndrome.
Frequency
Au-Kline syndrome is a rare condition with an unknown prevalence. At least 25 cases have been diagnosed.
Causes
Au-Kline syndrome is caused by mutations in the HNRNPK gene. The protein produced from this gene, called heterogenous nuclear ribonucleoprotein K (hnRNP K), attaches (binds) to DNA or its chemical cousin RNA and to other proteins, acting as a docking station for these molecules so they can interact. By bringing certain proteins together with DNA or RNA, the hnRNP K protein helps control the activity of genes and the production of proteins. By regulating certain genes and proteins, the hnRNP K protein plays a role in the normal development and function of several body systems, including the brain.
HNRNPK gene mutations that cause Au-Kline syndrome result in the production of little or no hnRNP K protein. A shortage of this protein alters gene activity and protein production, which disrupts the normal development or functioning of several body systems, resulting in the varied features of Au-Kline syndrome. In particular, problems with brain development likely contribute to intellectual disability, delayed development, and other neurological problems in people with the condition.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.

Most cases of this condition result from new (de novo) mutations in the gene that occur during the formation of reproductive cells (eggs or sperm) in an affected individual’s parent or in early embryonic development. These cases occur in people with no history of the disorder in their family.
Other Names for This Condition
- Okamoto syndrome
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Au PYB, Goedhart C, Ferguson M, Breckpot J, Devriendt K, Wierenga K, Fanning E, Grange DK, Graham GE, Galarreta C, Jones MC, Kini U, Stewart H, Parboosingh JS, Kline AD, Innes AM; Care for Rare Canada Consortium. Phenotypic spectrum of Au-Kline syndrome: a report of six new cases and review of the literature. Eur J Hum Genet. 2018 Sep;26(9):1272-1281. doi: 10.1038/s41431-018-0187-2. Epub 2018 Jun 14. Citation on PubMed or Free article on PubMed Central
- Au PYB, McNiven V, Phillips L, Innes AM, Kline AD. Au-Kline Syndrome. 2019 Apr 18 [updated 2024 Feb 1]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK540283/ Citation on PubMed
- Bomsztyk K, Denisenko O, Ostrowski J. hnRNP K: one protein multiple processes. Bioessays. 2004 Jun;26(6):629-38. doi: 10.1002/bies.20048. Citation on PubMed
- Folci A, Mapelli L, Sassone J, Prestori F, D'Angelo E, Bassani S, Passafaro M. Loss of hnRNP K impairs synaptic plasticity in hippocampal neurons. J Neurosci. 2014 Jul 2;34(27):9088-95. doi: 10.1523/JNEUROSCI.0303-14.2014. Citation on PubMed or Free article on PubMed Central
- Hutchins EJ, Szaro BG. c-Jun N-terminal kinase phosphorylation of heterogeneous nuclear ribonucleoprotein K regulates vertebrate axon outgrowth via a posttranscriptional mechanism. J Neurosci. 2013 Sep 11;33(37):14666-80. doi: 10.1523/JNEUROSCI.4821-12.2013. Citation on PubMed or Free article on PubMed Central
- Okamoto N, Matsumoto F, Shimada K, Satomura K. New MCA/MR syndrome with generalized hypotonia, congenital hydronephrosis, and characteristic face. Am J Med Genet. 1997 Jan 31;68(3):347-9. Citation on PubMed
- Okamoto N. Okamoto syndrome has features overlapping with Au-Kline syndrome and is caused by HNRNPK mutation. Am J Med Genet A. 2019 May;179(5):822-826. doi: 10.1002/ajmg.a.61079. Epub 2019 Feb 21. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.