

Description
Atypical hemolytic-uremic syndrome is a disease that primarily affects kidney function. This condition, which can occur at any age, causes abnormal blood clots (thrombi) to form in small blood vessels in the kidneys. These clots can cause serious medical problems if they restrict or block blood flow. Atypical hemolytic-uremic syndrome is characterized by three major features related to abnormal clotting: hemolytic anemia, thrombocytopenia, and kidney failure.
Hemolytic anemia occurs when red blood cells break down (undergo hemolysis) prematurely. In atypical hemolytic-uremic syndrome, red blood cells can break apart as they squeeze past clots within small blood vessels. Anemia results if these cells are destroyed faster than the body can replace them. Anemia can lead to unusually pale skin (pallor), yellowing of the eyes and skin (jaundice), fatigue, shortness of breath, and a rapid heart rate.
Thrombocytopenia is a reduced level of circulating platelets, which are cells that normally assist with blood clotting. In people with atypical hemolytic-uremic syndrome, fewer platelets are available in the bloodstream because a large number of platelets are used to make abnormal clots. Thrombocytopenia can cause easy bruising and abnormal bleeding.
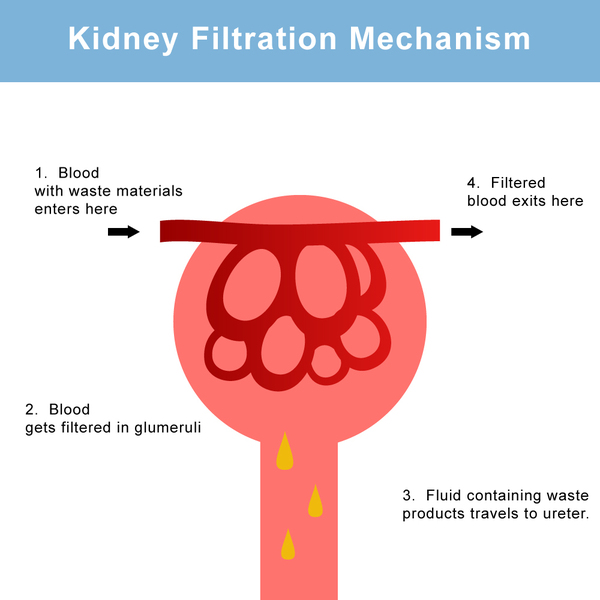
As a result of clot formation in small blood vessels, people with atypical hemolytic-uremic syndrome experience kidney damage and acute kidney failure that lead to end-stage renal disease (ESRD) in about half of all cases. These life-threatening complications prevent the kidneys from filtering fluids and waste products from the body effectively.
Atypical hemolytic-uremic syndrome should be distinguished from a more common condition called typical hemolytic-uremic syndrome. The two disorders have different causes and different signs and symptoms. Unlike the atypical form, the typical form is caused by infection with certain strains of Escherichia coli bacteria that produce toxic substances called Shiga-like toxins. The typical form is characterized by severe diarrhea and most often affects children younger than 10. The typical form is less likely than the atypical form to involve recurrent attacks of kidney damage that lead to ESRD.
Frequency
The incidence of atypical hemolytic-uremic syndrome is estimated to be 1 in 500,000 people per year in the United States. The atypical form is probably about 10 times less common than the typical form.
Causes
Atypical hemolytic-uremic syndrome often results from a combination of environmental and genetic factors. Mutations in at least seven genes appear to increase the risk of developing the disorder. Mutations in a gene called CFH are most common; they have been found in about 30 percent of all cases of atypical hemolytic-uremic syndrome. Mutations in the other genes have each been identified in a smaller percentage of cases.
The genes associated with atypical hemolytic-uremic syndrome provide instructions for making proteins involved in a part of the body's immune response known as the complement system. This system is a group of proteins that work together to destroy foreign invaders (such as bacteria and viruses), trigger inflammation, and remove debris from cells and tissues. The complement system must be carefully regulated so it targets only unwanted materials and does not attack the body's healthy cells. The regulatory proteins associated with atypical hemolytic-uremic syndrome protect healthy cells by preventing activation of the complement system when it is not needed.
Mutations in the genes associated with atypical hemolytic-uremic syndrome lead to uncontrolled activation of the complement system. The overactive system attacks cells that line blood vessels in the kidneys, causing inflammation and the formation of abnormal clots. These abnormalities lead to kidney damage and, in many cases, kidney failure and ESRD.
Although gene mutations increase the risk of atypical hemolytic-uremic syndrome, studies suggest that they are often not sufficient to cause the disease. In people with certain genetic changes, the signs and symptoms of the disorder may be triggered by factors including certain medications (such as anticancer drugs), chronic diseases, viral or bacterial infections, cancers, organ transplantation, or pregnancy.
Some people with atypical hemolytic-uremic syndrome do not have any known genetic changes or environmental triggers for the disease. In these cases, the disorder is described as idiopathic.
Inheritance
Most cases of atypical hemolytic-uremic syndrome are sporadic, which means that they occur in people with no apparent history of the disorder in their family. Less than 20 percent of all cases have been reported to run in families. When the disorder is familial, it can have an autosomal dominant or an autosomal recessive pattern of inheritance.
Autosomal dominant inheritance means one copy of an altered gene in each cell is sufficient to increase the risk of the disorder. In some cases, an affected person inherits the mutation from one affected parent. However, most people with the autosomal dominant form of atypical hemolytic-uremic syndrome have no history of the disorder in their family. Because not everyone who inherits a gene mutation will develop the signs and symptoms of the disease, an affected individual may have unaffected relatives who carry a copy of the mutation.

Autosomal recessive inheritance means both copies of a gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- AHUS
- Non-Shiga-like toxin-associated HUS
- Non-Stx-HUS
- Nonenteropathic HUS
Additional Information & Resources
Genetic Testing Information
- Genetic Testing Registry: Atypical hemolytic-uremic syndrome

- Genetic Testing Registry: Atypical hemolytic-uremic syndrome with B factor anomaly
- Genetic Testing Registry: Atypical hemolytic-uremic syndrome with C3 anomaly
- Genetic Testing Registry: Atypical hemolytic-uremic syndrome with I factor anomaly
- Genetic Testing Registry: Atypical hemolytic-uremic syndrome with MCP/CD46 anomaly
- Genetic Testing Registry: Atypical hemolytic-uremic syndrome with thrombomodulin anomaly
- Genetic Testing Registry: Hemolytic uremic syndrome, atypical, susceptibility to, 1
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
- HEMOLYTIC UREMIC SYNDROME, ATYPICAL, SUSCEPTIBILITY TO, 1; AHUS1
- HEMOLYTIC UREMIC SYNDROME, ATYPICAL, SUSCEPTIBILITY TO, 2; AHUS2
- HEMOLYTIC UREMIC SYNDROME, ATYPICAL, SUSCEPTIBILITY TO, 3; AHUS3
- HEMOLYTIC UREMIC SYNDROME, ATYPICAL, SUSCEPTIBILITY TO, 4; AHUS4
- HEMOLYTIC UREMIC SYNDROME, ATYPICAL, SUSCEPTIBILITY TO, 5; AHUS5
- HEMOLYTIC UREMIC SYNDROME, ATYPICAL, SUSCEPTIBILITY TO, 6; AHUS6
Scientific Articles on PubMed
References
- Caprioli J, Noris M, Brioschi S, Pianetti G, Castelletti F, Bettinaglio P, Mele C, Bresin E, Cassis L, Gamba S, Porrati F, Bucchioni S, Monteferrante G, Fang CJ, Liszewski MK, Kavanagh D, Atkinson JP, Remuzzi G; International Registry of Recurrent and Familial HUS/TTP. Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood. 2006 Aug 15;108(4):1267-79. doi: 10.1182/blood-2005-10-007252. Epub 2006 Apr 18. Citation on PubMed or Free article on PubMed Central
- Hirt-Minkowski P, Dickenmann M, Schifferli JA. Atypical hemolytic uremic syndrome: update on the complement system and what is new. Nephron Clin Pract. 2010;114(4):c219-35. doi: 10.1159/000276545. Epub 2010 Jan 14. Citation on PubMed
- Jokiranta TS, Zipfel PF, Fremeaux-Bacchi V, Taylor CM, Goodship TJ, Noris M. Where next with atypical hemolytic uremic syndrome? Mol Immunol. 2007 Sep;44(16):3889-900. doi: 10.1016/j.molimm.2007.06.003. Citation on PubMed
- Loirat C, Noris M, Fremeaux-Bacchi V. Complement and the atypical hemolytic uremic syndrome in children. Pediatr Nephrol. 2008 Nov;23(11):1957-72. doi: 10.1007/s00467-008-0872-4. Epub 2008 Jul 2. Citation on PubMed
- Noris M, Bresin E, Mele C, Remuzzi G. Genetic Atypical Hemolytic-Uremic Syndrome. 2007 Nov 16 [updated 2021 Sep 23]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1367/ Citation on PubMed
- Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med. 2009 Oct 22;361(17):1676-87. doi: 10.1056/NEJMra0902814. No abstract available. Citation on PubMed
- Noris M, Remuzzi G. Hemolytic uremic syndrome. J Am Soc Nephrol. 2005 Apr;16(4):1035-50. doi: 10.1681/ASN.2004100861. Epub 2005 Feb 23. No abstract available. Citation on PubMed
- Sellier-Leclerc AL, Fremeaux-Bacchi V, Dragon-Durey MA, Macher MA, Niaudet P, Guest G, Boudailliez B, Bouissou F, Deschenes G, Gie S, Tsimaratos M, Fischbach M, Morin D, Nivet H, Alberti C, Loirat C; French Society of Pediatric Nephrology. Differential impact of complement mutations on clinical characteristics in atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2007 Aug;18(8):2392-400. doi: 10.1681/ASN.2006080811. Epub 2007 Jun 28. Citation on PubMed
- Sullivan M, Erlic Z, Hoffmann MM, Arbeiter K, Patzer L, Budde K, Hoppe B, Zeier M, Lhotta K, Rybicki LA, Bock A, Berisha G, Neumann HP. Epidemiological approach to identifying genetic predispositions for atypical hemolytic uremic syndrome. Ann Hum Genet. 2010 Jan;74(1):17-26. doi: 10.1111/j.1469-1809.2009.00554.x. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.