

Description
Atelosteogenesis type 2 is a severe disorder of cartilage and bone development. Infants born with this condition have very short arms and legs, a narrow chest, and a prominent, rounded abdomen. This disorder is also characterized by an opening in the roof of the mouth (a cleft palate), distinctive facial features, an inward- and upward-turning foot (clubfoot), and unusually positioned thumbs (hitchhiker thumbs).


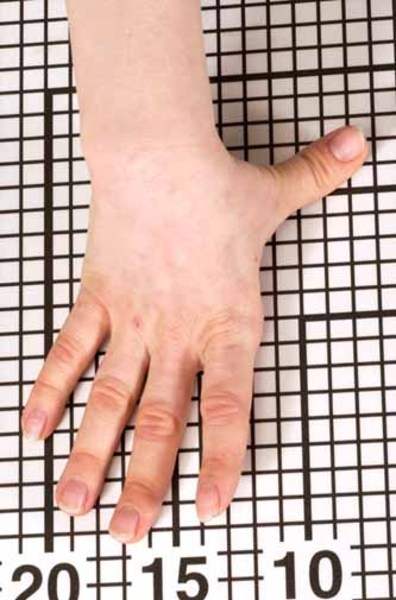
The signs and symptoms of atelosteogenesis type 2 are similar to those of another skeletal disorder called diastrophic dysplasia; however, atelosteogenesis type 2 is typically more severe. As a result of serious health problems, infants with this disorder are usually stillborn or die soon after birth from respiratory failure. Some infants, however, have lived for a short time with intensive medical support.
Frequency
Atelosteogenesis type 2 is an extremely rare genetic disorder; its incidence is unknown.
Causes
Atelosteogenesis type 2 is one of several skeletal disorders caused by mutations in the SLC26A2 gene. This gene provides instructions for making a protein that is essential for the normal development of cartilage and for its conversion to bone. Cartilage is a tough, flexible tissue that makes up much of the skeleton during early development. Most cartilage is later converted to bone, except for the cartilage that continues to cover and protect the ends of bones and is present in the nose and external ears. Mutations in the SLC26A2 gene disrupt the structure of developing cartilage, preventing bones from forming properly and resulting in the skeletal problems characteristic of atelosteogenesis type 2.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- AO2
- Atelosteogenesis de la Chapelle type
- Atelosteogenesis, type 2
- De la Chapelle dysplasia
- McAlister dysplasia
- Neonatal osseous dysplasia 1
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Hastbacka J, Superti-Furga A, Wilcox WR, Rimoin DL, Cohn DH, Lander ES. Atelosteogenesis type II is caused by mutations in the diastrophic dysplasia sulfate-transporter gene (DTDST): evidence for a phenotypic series involving three chondrodysplasias. Am J Hum Genet. 1996 Feb;58(2):255-62. Citation on PubMed or Free article on PubMed Central
- Newbury-Ecob R. Atelosteogenesis type 2. J Med Genet. 1998 Jan;35(1):49-53. doi: 10.1136/jmg.35.1.49. Citation on PubMed or Free article on PubMed Central
- Rossi A, Superti-Furga A. Mutations in the diastrophic dysplasia sulfate transporter (DTDST) gene (SLC26A2): 22 novel mutations, mutation review, associated skeletal phenotypes, and diagnostic relevance. Hum Mutat. 2001 Mar;17(3):159-71. doi: 10.1002/humu.1. Erratum In: Hum Mutat 2001;18(1):82. Citation on PubMed
- Superti-Furga A, Unger S. SLC26A2-Related Atelosteogenesis. 2002 Aug 30 [updated 2023 Mar 16]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1317/ Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.