

Description
Atelosteogenesis type 1 is a disorder that affects the development of bones throughout the body. Affected individuals are born with inward- and upward-turning feet (clubfeet) and dislocations of the hips, knees, and elbows. Bones in the spine, rib cage, pelvis, and limbs may be underdeveloped or in some cases absent. As a result of the limb bone abnormalities, individuals with this condition have very short arms and legs. Characteristic facial features include a prominent forehead, wide-set eyes (hypertelorism), an upturned nose with a grooved tip, and a very small lower jaw and chin (micrognathia). Affected individuals may also have an opening in the roof of the mouth (a cleft palate). Males with this condition can have undescended testes.

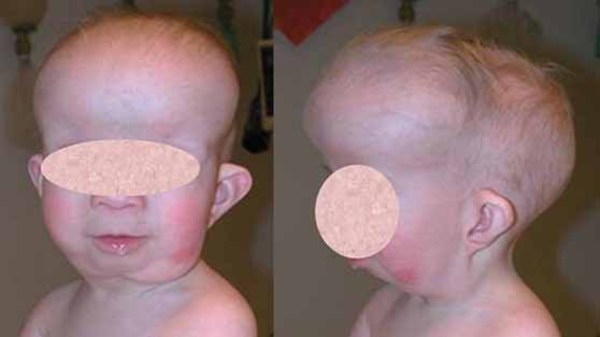

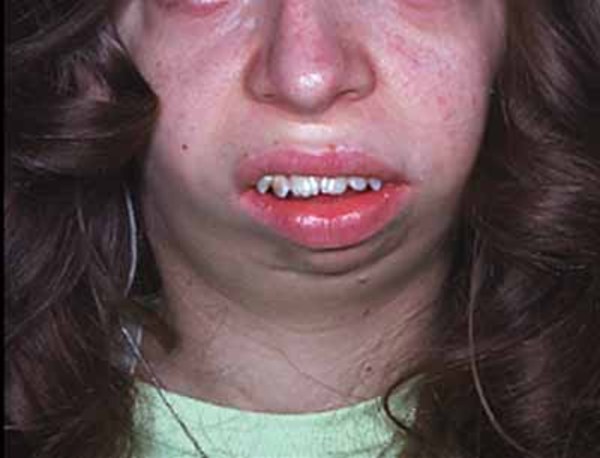

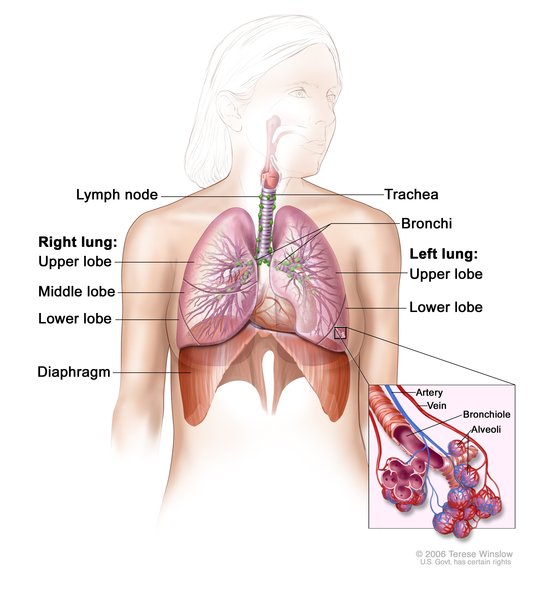
Individuals with atelosteogenesis type 1 typically have an underdeveloped rib cage that affects the development and functioning of the lungs. As a result, affected individuals are usually stillborn or die shortly after birth from respiratory failure.
Frequency
Atelosteogenesis type 1 is a rare disorder; its exact prevalence is unknown. Only a few dozen affected individuals have been identified.
Causes
Mutations in the FLNB gene cause atelosteogenesis type 1. The FLNB gene provides instructions for making a protein called filamin B. This protein helps build the network of protein filaments (cytoskeleton) that gives structure to cells and allows them to change shape and move. Filamin B attaches (binds) to another protein called actin and helps the actin to form the branching network of filaments that makes up the cytoskeleton. Filamin B also links actin to many other proteins to perform various functions within the cell, including the cell signaling that helps determine how the cytoskeleton will change as tissues grow and take shape during development.

Filamin B is especially important in the development of the skeleton before birth. It is active (expressed) in the cell membranes of cartilage-forming cells (chondrocytes). Cartilage is a tough, flexible tissue that makes up much of the skeleton during early development. Most cartilage is later converted to bone, a process called ossification, except for the cartilage that continues to cover and protect the ends of bones and is present in the nose, airways (trachea and bronchi), and external ears. Filamin B appears to be important for normal cell growth and division (proliferation) and maturation (differentiation) of chondrocytes and for the ossification of cartilage.
FLNB gene mutations that cause atelosteogenesis type 1 change single protein building blocks (amino acids) in the filamin B protein or delete a small section of the protein sequence, resulting in an abnormal protein. This abnormal protein appears to have a new, atypical function that interferes with the proliferation or differentiation of chondrocytes, impairing ossification and leading to the signs and symptoms of atelosteogenesis type 1.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. Almost all cases result from new mutations in the gene and occur in people with no history of the disorder in their family.

Other Names for This Condition
- AOI
- Atelosteogenesis type I
- Giant cell chondrodysplasia
- Spondylohumerofemoral hypoplasia
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Farrington-Rock C, Firestein MH, Bicknell LS, Superti-Furga A, Bacino CA, Cormier-Daire V, Le Merrer M, Baumann C, Roume J, Rump P, Verheij JB, Sweeney E, Rimoin DL, Lachman RS, Robertson SP, Cohn DH, Krakow D. Mutations in two regions of FLNB result in atelosteogenesis I and III. Hum Mutat. 2006 Jul;27(7):705-10. doi: 10.1002/humu.20348. Citation on PubMed
- Krakow D, Robertson SP, King LM, Morgan T, Sebald ET, Bertolotto C, Wachsmann-Hogiu S, Acuna D, Shapiro SS, Takafuta T, Aftimos S, Kim CA, Firth H, Steiner CE, Cormier-Daire V, Superti-Furga A, Bonafe L, Graham JM Jr, Grix A, Bacino CA, Allanson J, Bialer MG, Lachman RS, Rimoin DL, Cohn DH. Mutations in the gene encoding filamin B disrupt vertebral segmentation, joint formation and skeletogenesis. Nat Genet. 2004 Apr;36(4):405-10. doi: 10.1038/ng1319. Epub 2004 Feb 29. Citation on PubMed
- Sawyer GM, Clark AR, Robertson SP, Sutherland-Smith AJ. Disease-associated substitutions in the filamin B actin binding domain confer enhanced actin binding affinity in the absence of major structural disturbance: Insights from the crystal structures of filamin B actin binding domains. J Mol Biol. 2009 Jul 31;390(5):1030-47. doi: 10.1016/j.jmb.2009.06.009. Epub 2009 Jun 6. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.