

Description
Aspartylglucosaminuria is a condition that primarily affects mental functioning and movement. This conditions worsens over time. Infants with aspartylglucosaminuria appear healthy at birth, and development is typically normal throughout early childhood. Around the age of 2 or 3, affected children usually begin to have delayed speech, mild intellectual disability, and problems coordinating movements. Other features that develop in childhood include respiratory infections, a protrusion of organs through gaps in muscles (hernia), and a growth spurt resulting in a large head size (macrocephaly).
Intellectual disability and movement problems worsen in adolescence. Most people with this disorder lose much of the speech they have learned, and affected adults usually have only a few words in their vocabulary. Adults with aspartylglucosaminuria often have psychological disorders and may develop seizures.
People with aspartylglucosaminuria may also have bones that become progressively weak and prone to fracture (osteoporosis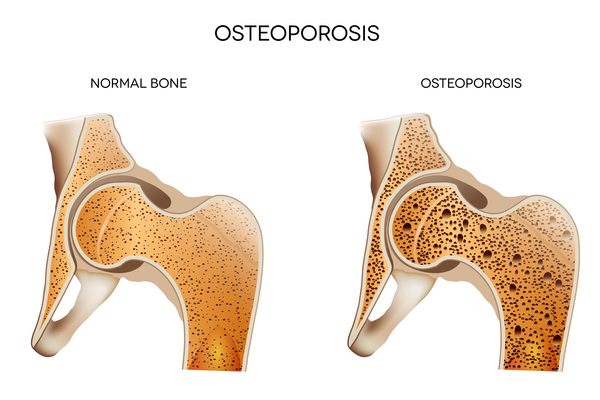 ), an unusually large range of joint movement (hypermobility), and loose skin. Affected individuals tend to have a characteristic facial appearance that includes widely spaced eyes (ocular hypertelorism
), an unusually large range of joint movement (hypermobility), and loose skin. Affected individuals tend to have a characteristic facial appearance that includes widely spaced eyes (ocular hypertelorism ), small ears
), small ears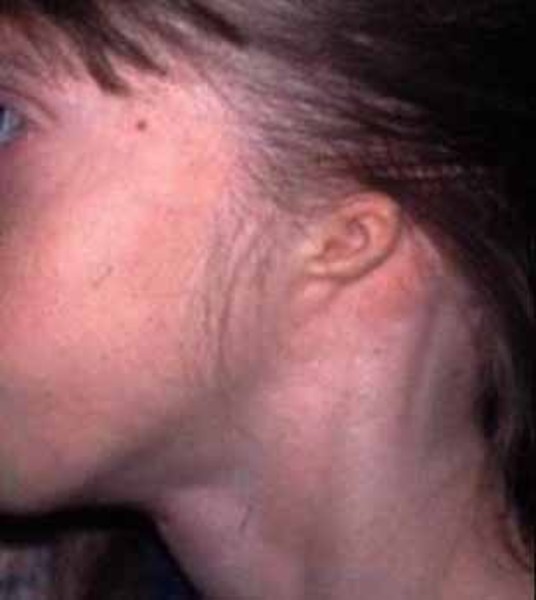 , and full lips. The nose is short and broad
, and full lips. The nose is short and broad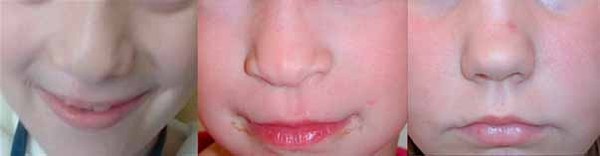 and the face is usually square-shaped. They often have poor oral health, including infections and gum disease (gingivitis). Children with this condition may be tall for their age, but lack of a growth spurt in puberty typically causes adults to be short with a small head size (microcephaly). Individuals with aspartylglucosaminuria usually survive into mid-adulthood.
and the face is usually square-shaped. They often have poor oral health, including infections and gum disease (gingivitis). Children with this condition may be tall for their age, but lack of a growth spurt in puberty typically causes adults to be short with a small head size (microcephaly). Individuals with aspartylglucosaminuria usually survive into mid-adulthood.
Frequency
In Finland, it is estimated that 1 to 3 individuals are born with aspartylglucosaminuria each year. This condition is less common outside of Finland, but the incidence is unknown.
Causes
Variants (also known as mutations) in the AGA gene cause aspartylglucosaminuria. The AGA gene provides instructions for producing an enzyme called aspartylglucosaminidase. This enzyme is active in lysosomes , which are structures inside cells that act as recycling centers. Within lysosomes, the enzyme helps break down complex chains of sugar molecules (oligosaccharides) attached to certain proteins (glycoproteins).
, which are structures inside cells that act as recycling centers. Within lysosomes, the enzyme helps break down complex chains of sugar molecules (oligosaccharides) attached to certain proteins (glycoproteins).
AGA gene variants result in a lack (deficiency) of the aspartylglucosaminidase enzyme in lysosomes, preventing the normal breakdown of glycoproteins. As a result, glycoproteins can build up within the lysosomes. Excess glycoproteins disrupt the normal functions of the cell and can result in cell death. A buildup of glycoproteins seems to particularly affect nerve cells in the brain; loss of these cells causes many of the signs and symptoms of aspartylglucosaminuria.
Inheritance
This condition is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell have variants. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
, which means both copies of the gene in each cell have variants. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
Other Names for This Condition
- AGA deficiency
- Aspartylglucosamidase deficiency
- Aspartylglucosaminidase deficiency
- Aspartylglycosaminuria
- Glycosylasparaginase deficiency
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Aronson NN Jr. Aspartylglycosaminuria: biochemistry and molecular biology. Biochim Biophys Acta. 1999 Oct 8;1455(2-3):139-54. doi: 10.1016/s0925-4439(99)00076-9. Citation on PubMed
- Arvio M, Mononen I. Aspartylglycosaminuria: a review. Orphanet J Rare Dis. 2016 Dec 1;11(1):162. doi: 10.1186/s13023-016-0544-6. Citation on PubMed
- Arvio MA, Peippo MM, Arvio PJ, Kaariainen HA. Dysmorphic facial features in aspartylglucosaminuria patients and carriers. Clin Dysmorphol. 2004 Jan;13(1):11-5. doi: 10.1097/00019605-200401000-00003. Citation on PubMed
- Goodspeed K, Feng C, Laine M, Lund TC. Aspartylglucosaminuria: Clinical Presentation and Potential Therapies. J Child Neurol. 2021 Apr;36(5):403-414. doi: 10.1177/0883073820980904. Epub 2021 Jan 13. Citation on PubMed
- Saarela J, Laine M, Oinonen C, von Schantz C, Jalanko A, Rouvinen J, Peltonen L. Molecular pathogenesis of a disease: structural consequences of aspartylglucosaminuria mutations. Hum Mol Genet. 2001 Apr 15;10(9):983-95. doi: 10.1093/hmg/10.9.983. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.