

Description
Ankyloblepharon-ectodermal defects-cleft lip/palate (AEC) syndrome is a form of ectodermal dysplasia, a group of about 180 conditions characterized by abnormal development of ectodermal tissues including the skin, hair, nails, teeth, eyes, ears, and sweat glands.
Among the most common features of AEC syndrome are missing patches of skin (erosion). In affected infants, skin erosion most commonly occurs on the scalp. It tends to recur throughout childhood and into adulthood, frequently affecting the scalp, neck, hands, and feet. Skin erosion ranges from mild to severe and can lead to life-threatening infection in infancy, scarring, and hair loss. Other ectodermal abnormalities in AEC syndrome include changes in skin coloring; brittle, sparse, or missing hair; misshapen or absent fingernails and toenails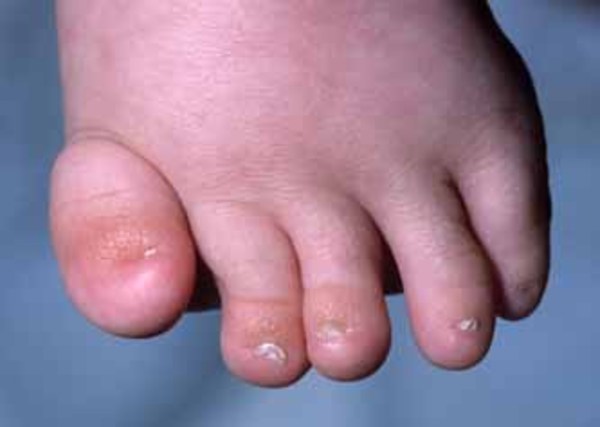 ; and malformed or missing teeth. Affected individuals may also have an inability to control their body temperature because of missing or nonfunctioning sweat glands causing overheating or hypothermia.
; and malformed or missing teeth. Affected individuals may also have an inability to control their body temperature because of missing or nonfunctioning sweat glands causing overheating or hypothermia.
Many infants with AEC syndrome are born with an eyelid condition known as ankyloblepharon filiforme adnatum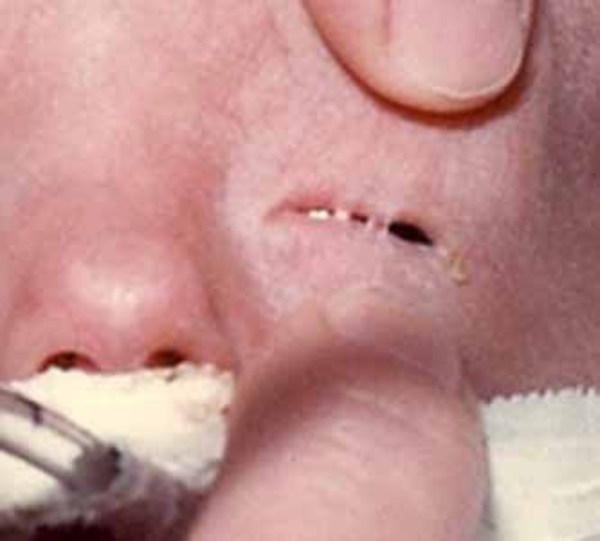 , in which strands of tissue partially or completely fuse the upper and lower eyelids. Most people with AEC syndrome are also born with an opening in the roof of the mouth (a cleft palate
, in which strands of tissue partially or completely fuse the upper and lower eyelids. Most people with AEC syndrome are also born with an opening in the roof of the mouth (a cleft palate ), a split in the lip (a cleft lip
), a split in the lip (a cleft lip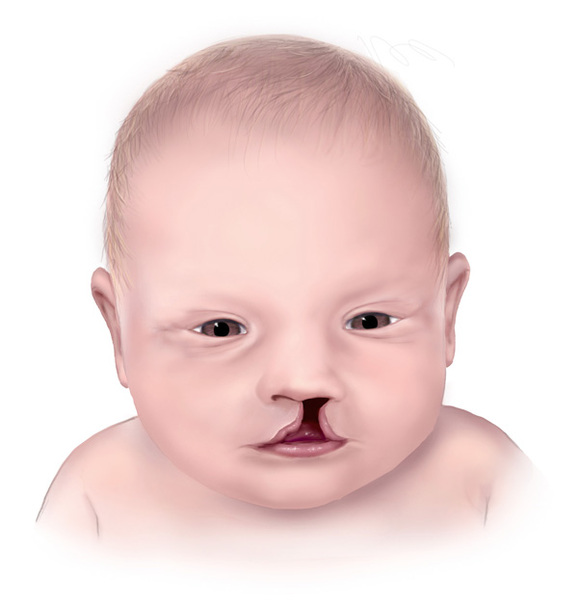 ), or both. Cleft lip or cleft palate can make it difficult for affected infants to suck, so these infants often have trouble feeding and do not grow and gain weight at the expected rate (failure to thrive).
), or both. Cleft lip or cleft palate can make it difficult for affected infants to suck, so these infants often have trouble feeding and do not grow and gain weight at the expected rate (failure to thrive).
Additional features of AEC syndrome can include limb abnormalities, most commonly fused fingers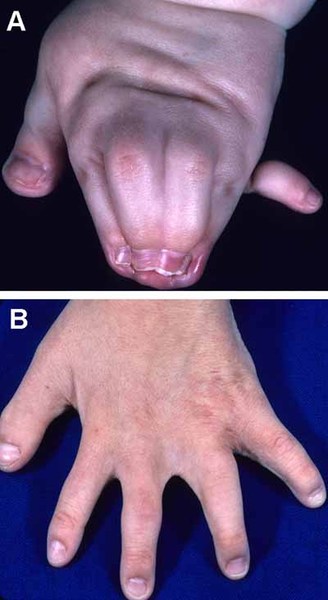 and toes
and toes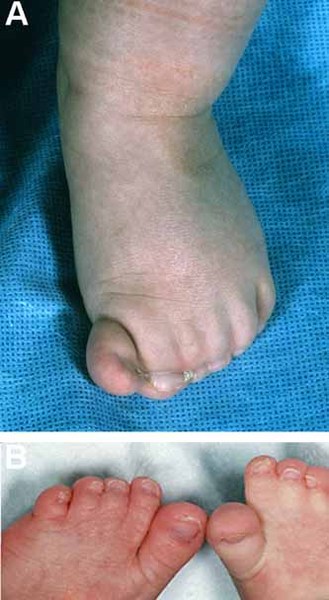 (syndactyly). Less often, affected individuals have permanently bent fingers and toes (camptodactyly
(syndactyly). Less often, affected individuals have permanently bent fingers and toes (camptodactyly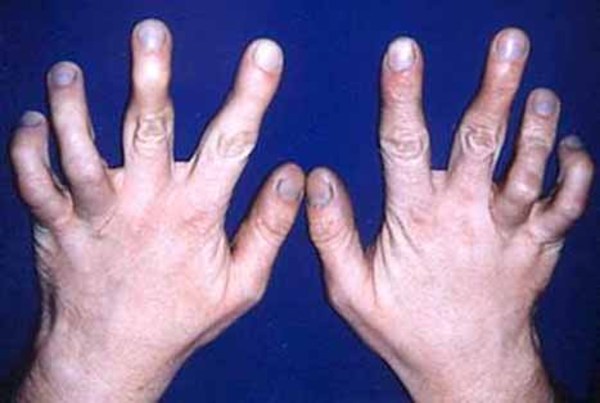 ) or a deep split in the hands
) or a deep split in the hands or feet
or feet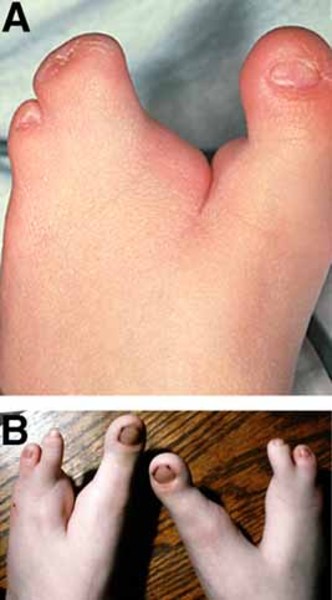 with missing fingers or toes and fusion of the remaining digits (ectrodactyly). Hearing loss is common, occurring in more than 90 percent of children with AEC syndrome. Some affected individuals have distinctive facial features, such as small jaws that cannot open fully and a narrow space between the upper lip and nose (philtrum). Other signs and symptoms can include the opening of the urethra on the underside of the penis (hypospadias) in affected males, digestive problems, absent tear duct openings in the eyes, and chronic sinus or ear infections.
with missing fingers or toes and fusion of the remaining digits (ectrodactyly). Hearing loss is common, occurring in more than 90 percent of children with AEC syndrome. Some affected individuals have distinctive facial features, such as small jaws that cannot open fully and a narrow space between the upper lip and nose (philtrum). Other signs and symptoms can include the opening of the urethra on the underside of the penis (hypospadias) in affected males, digestive problems, absent tear duct openings in the eyes, and chronic sinus or ear infections.
A condition known as Rapp-Hodgkin syndrome has signs and symptoms that overlap considerably with those of AEC syndrome. These two syndromes were classified as separate disorders until it was discovered that they both result from mutations in the same part of the same gene. Most researchers now consider Rapp-Hodgkin syndrome and AEC syndrome to be part of the same disease spectrum.
Frequency
AEC syndrome is a rare condition; its prevalence is unknown. All forms of ectodermal dysplasia together occur in about 1 in 70,000 newborns worldwide.
Causes
AEC syndrome is caused by variants (also known as mutations) in the TP63 gene. This gene provides instructions for making a protein known as p63, which plays an essential role in early development. The p63 protein is a transcription factor, which means that it attaches (binds) to DNA and controls the activity of particular genes. The p63 protein turns many different genes on and off during development. It appears to be especially critical for the development of ectodermal structures, such as the skin, hair, teeth, and nails. Studies suggest that it also plays important roles in the development of the limbs, facial features, urinary system, and other organs and tissues.
The TP63 gene variants responsible for AEC syndrome interfere with the ability of p63 to turn target genes on and off at the right times. It is unclear how these changes lead to abnormal ectodermal development and the specific features of AEC syndrome.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder.

Other Names for This Condition
- AEC syndrome
- Ankyloblepharon-ectodermal defects-cleft lip and palate syndrome
- Hay-Wells syndrome
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Clements SE, Techanukul T, Holden ST, Mellerio JE, Dorkins H, Escande F, McGrath JA. Rapp-Hodgkin and Hay-Wells ectodermal dysplasia syndromes represent a variable spectrum of the same genetic disorder. Br J Dermatol. 2010 Sep;163(3):624-9. doi: 10.1111/j.1365-2133.2010.09859.x. Citation on PubMed
- Cole P, Hatef DA, Kaufman Y, Magruder A, Bree A, Friedman E, Sindwani R, Hollier LH Jr. Facial clefting and oroauditory pathway manifestations in ankyloblepharon-ectodermal defects-cleft lip/palate (AEC) syndrome. Am J Med Genet A. 2009 Sep;149A(9):1910-5. doi: 10.1002/ajmg.a.32836. Citation on PubMed
- Dishop MK, Bree AF, Hicks MJ. Pathologic changes of skin and hair in ankyloblepharon-ectodermal defects-cleft lip/palate (AEC) syndrome. Am J Med Genet A. 2009 Sep;149A(9):1935-41. doi: 10.1002/ajmg.a.32826. Citation on PubMed
- Farrington F, Lausten L. Oral findings in ankyloblepharon-ectodermal dysplasia-cleft lip/palate (AEC) syndrome. Am J Med Genet A. 2009 Sep;149A(9):1907-9. doi: 10.1002/ajmg.a.32790. Citation on PubMed
- Julapalli MR, Scher RK, Sybert VP, Siegfried EC, Bree AF. Dermatologic findings of ankyloblepharon-ectodermal defects-cleft lip/palate (AEC) syndrome. Am J Med Genet A. 2009 Sep;149A(9):1900-6. doi: 10.1002/ajmg.a.32797. Citation on PubMed
- McGrath JA, Duijf PH, Doetsch V, Irvine AD, de Waal R, Vanmolkot KR, Wessagowit V, Kelly A, Atherton DJ, Griffiths WA, Orlow SJ, van Haeringen A, Ausems MG, Yang A, McKeon F, Bamshad MA, Brunner HG, Hamel BC, van Bokhoven H. Hay-Wells syndrome is caused by heterozygous missense mutations in the SAM domain of p63. Hum Mol Genet. 2001 Feb 1;10(3):221-9. doi: 10.1093/hmg/10.3.221. Citation on PubMed
- Motil KJ, Fete TJ. Growth, nutritional, and gastrointestinal aspects of ankyloblepharon-ectodermal defect-cleft lip and/or palate (AEC) syndrome. Am J Med Genet A. 2009 Sep;149A(9):1922-5. doi: 10.1002/ajmg.a.32789. Citation on PubMed
- Siegfried E, Bree A, Fete M, Sybert VP. Skin erosions and wound healing in ankyloblepharon-ectodermal defect-cleft lip and/or palate. Arch Dermatol. 2005 Dec;141(12):1591-4. doi: 10.1001/archderm.141.12.1591. No abstract available. Citation on PubMed
- Sutton VR, Plunkett K, Dang DX, Lewis RA, Bree AF, Bacino CA. Craniofacial and anthropometric phenotype in ankyloblepharon-ectodermal defects-cleft lip/palate syndrome (Hay-Wells syndrome) in a cohort of 17 patients. Am J Med Genet A. 2009 Sep;149A(9):1916-21. doi: 10.1002/ajmg.a.32791. Citation on PubMed
- Sutton VR, van Bokhoven H. TP63-Related Disorders. 2010 Jun 8 [updated 2021 Apr 1]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK43797/ Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.