

Description
Andersen-Tawil syndrome is a disorder that causes episodes of muscle weakness (periodic paralysis), changes in heart rhythm (arrhythmia), and developmental abnormalities. Periodic paralysis begins early in life, and episodes last from hours to days. These episodes may occur after exercise or long periods of rest, but they often have no obvious trigger. Muscle strength usually returns to normal between episodes. However, mild muscle weakness may eventually become permanent.
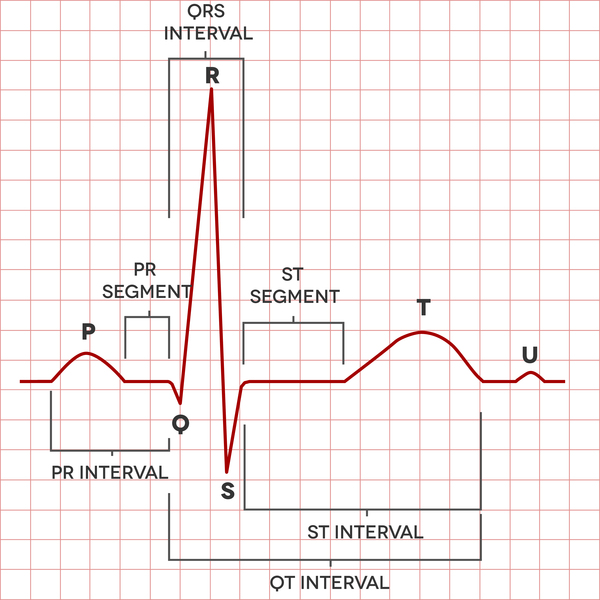
In people with Andersen-Tawil syndrome, the most common changes affecting the heart are ventricular arrhythmia, which is a disruption in the rhythm of the heart's lower chambers (the ventricles), and long QT syndrome. Long QT syndrome is a heart condition that causes the heart (cardiac) muscle to take longer than usual to recharge between beats. The irregular heartbeats can lead to discomfort, such as the feeling that the heart is skipping beats (palpitations). Uncommonly, the irregular heartbeats can cause fainting (syncope), and even more rarely, sudden death.
Physical abnormalities associated with Andersen-Tawil syndrome typically affect the face, other parts of the head, and the limbs. These features often include a very small lower jaw (micrognathia), dental abnormalities (such as crowded teeth), low-set ears, widely spaced eyes, fusion (syndactyly) of the second and third toes, and unusual curving of the fingers or toes (clinodactyly). Some affected people also have short stature and an abnormal side-to-side curvature of the spine (scoliosis).
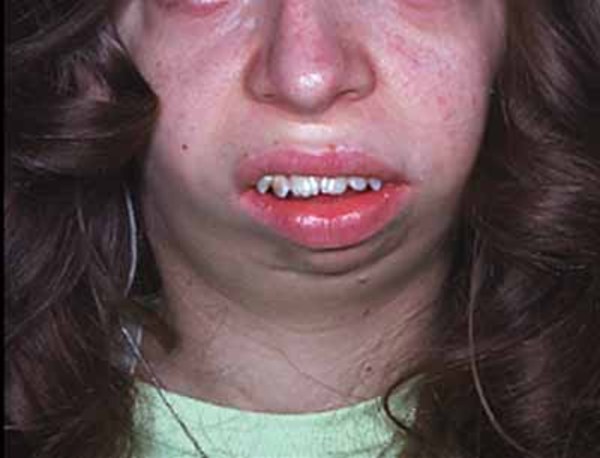

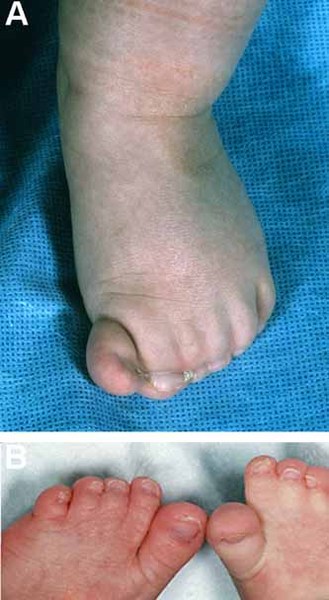
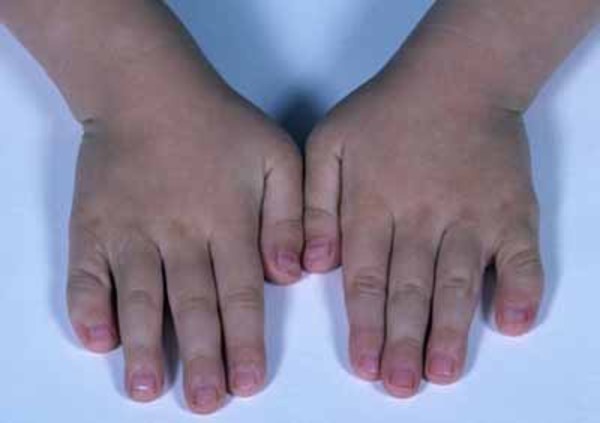

The signs and symptoms of Andersen-Tawil syndrome vary widely, and they can be different even among affected members of the same family. About 60 percent of affected individuals have all three major features (periodic paralysis, cardiac arrhythmia, and physical abnormalities).
Frequency
Andersen-Tawil syndrome is a rare genetic disorder. Its exact prevalence is unknown, although it is estimated to affect 1 in 1 million people worldwide. About 200 affected individuals have been described in the medical literature. Researchers believe that Andersen-Tawil syndrome accounts for less than 10 percent of all cases of periodic paralysis.
Causes
Mutations in the KCNJ2 gene cause about 60 percent of all cases of Andersen-Tawil syndrome. When the disorder is caused by mutations in this gene, it is classified as type 1 (ATS1).
The KCNJ2 gene provides instructions for making channels that transport positively charged potassium ions across the membrane of muscle cells. The movement of potassium ions through these channels is critical for maintaining the normal function of muscles used for movement (skeletal muscles) and cardiac muscle. Mutations in the KCNJ2 gene alter the usual structure and function of these potassium channels. These changes disrupt the flow of potassium ions in skeletal and cardiac muscle, leading to the periodic paralysis and irregular heart rhythm characteristic of Andersen-Tawil syndrome.

Researchers have not determined the role of the KCNJ2 gene in bone development, and it is not known how mutations in the gene lead to the skeletal changes and other physical abnormalities often found in Andersen-Tawil syndrome.
In the 40 percent of cases not caused by KCNJ2 gene mutations, the cause of Andersen-Tawil syndrome is usually unknown. These cases are classified as type 2 (ATS2). Studies suggest that variations in at least one other potassium channel gene may underlie the disorder in some of these affected individuals.
Inheritance
This condition is inherited in an autosomal dominant pattern, which means one copy of an altered gene in each cell is sufficient to cause the disorder. When the condition results from a mutation in the KCNJ2 gene, an affected individual may inherit the mutation from one affected parent. In other cases, the condition results from a new (de novo) mutation in the KCNJ2 gene. These cases occur in people with no history of the disorder in their family.


Other Names for This Condition
- Andersen syndrome
- ATS
- Long QT syndrome 7
- LQT7
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Ardissone A, Sansone V, Colleoni L, Bernasconi P, Moroni I. Intrafamilial phenotypic variability in Andersen-Tawil syndrome: A diagnostic challenge in a potentially treatable condition. Neuromuscul Disord. 2017 Mar;27(3):294-297. doi: 10.1016/j.nmd.2016.11.006. Epub 2016 Nov 18. Citation on PubMed
- Modell SM, Lehmann MH. The long QT syndrome family of cardiac ion channelopathies: a HuGE review. Genet Med. 2006 Mar;8(3):143-55. doi: 10.1097/01.gim.0000204468.85308.86. Citation on PubMed
- Nguyen HL, Pieper GH, Wilders R. Andersen-Tawil syndrome: clinical and molecular aspects. Int J Cardiol. 2013 Dec 5;170(1):1-16. doi: 10.1016/j.ijcard.2013.10.010. Citation on PubMed
- Plaster NM, Tawil R, Tristani-Firouzi M, Canun S, Bendahhou S, Tsunoda A, Donaldson MR, Iannaccone ST, Brunt E, Barohn R, Clark J, Deymeer F, George AL Jr, Fish FA, Hahn A, Nitu A, Ozdemir C, Serdaroglu P, Subramony SH, Wolfe G, Fu YH, Ptacek LJ. Mutations in Kir2.1 cause the developmental and episodic electrical phenotypes of Andersen's syndrome. Cell. 2001 May 18;105(4):511-9. doi: 10.1016/s0092-8674(01)00342-7. Citation on PubMed
- Tristani-Firouzi M, Etheridge SP. Kir 2.1 channelopathies: the Andersen-Tawil syndrome. Pflugers Arch. 2010 Jul;460(2):289-94. doi: 10.1007/s00424-010-0820-6. Epub 2010 Mar 21. Citation on PubMed
- Veerapandiyan A, Statland JM, Tawil R. Andersen-Tawil Syndrome. 2004 Nov 22 [updated 2018 Jun 7]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1264/ Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.