

Description
Alpha-N-acetylgalactosaminidase deficiency, also known as Schindler disease, is an inherited disorder that can affect the development of the nervous system. People with alpha-N-acetylgalactosaminidase deficiency often have distinctive facial features that can be described as "coarse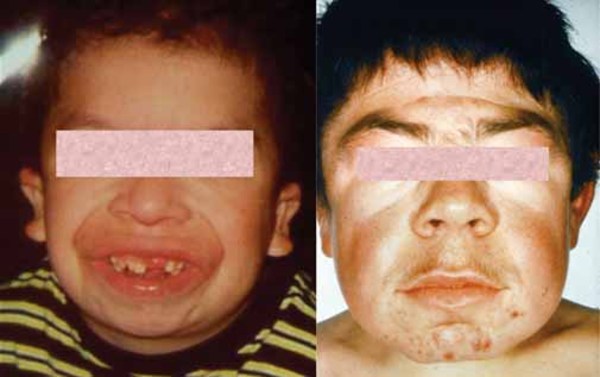 " and tooth abnormalities such as widely-spaced teeth and missing teeth (hypodontia).
" and tooth abnormalities such as widely-spaced teeth and missing teeth (hypodontia).
The three types of alpha-N-acetylgalactosaminidase deficiency differ in the severity of their signs and symptoms and the age at which they first appear. The signs and symptoms of alpha-N-acetylgalactosaminidase deficiency can vary, even among members of the same family.
Type I is the most severe form of this condition. Babies with alpha-N-acetylgalactosaminidase deficiency type I appear healthy at birth. However, by late infancy, these babies typically have trouble meeting normal developmental milestones. By the age of 2 years, individuals with type I begin to lose skills that they had already acquired (developmental regression). Individuals with type I often experience weak muscle tone (hypotonia), vision and hearing loss, and seizures. During early childhood, children with type I typically lose awareness of their surroundings and eventually become unresponsive.
Alpha-N-acetylgalactosaminidase deficiency type II is the least severe form. Type II is typically diagnosed in adulthood. Affected individuals may have mild cognitive impairment and hearing loss caused by abnormalities of the inner ear  (sensorineural hearing loss). They may also experience a loss of sensation or muscle weakness in the extremities (peripheral neuropathy). Clusters of enlarged blood vessels that form small, dark red spots on the skin (angiokeratomas) are a characteristic feature of alpha-N-acetylgalactosaminidase deficiency type II.
(sensorineural hearing loss). They may also experience a loss of sensation or muscle weakness in the extremities (peripheral neuropathy). Clusters of enlarged blood vessels that form small, dark red spots on the skin (angiokeratomas) are a characteristic feature of alpha-N-acetylgalactosaminidase deficiency type II.
Type III is the intermediate form of alpha-N-acetylgalactosaminidase deficiency. People with type III may show a variety of signs and symptoms, including developmental, speech, and language delays; seizures that begin in infancy; and features of autism spectrum disorder that appear in childhood. Autism spectrum disorder is characterized by impaired communication and socialization skills. People with type III may also have skeletal signs and symptoms, such as pain in the lower back, hips, and knees. Wear on the cartilage (disks) and bones of the neck (cervical spondylosis) and a cyst-like collection of cerebrospinal fluid that forms in the spinal cord (syringohydromyelia) have also been reported.
Frequency
Alpha-N-acetylgalactosaminidase deficiency is very rare. Fewer than 50 individuals with this condition have been reported in the medical literature. Some cases of alpha-N-acetylgalactosaminidase deficiency likely remain undiagnosed.
Causes
Variants (also called mutations) in the NAGA gene cause alpha-N-acetylgalactosaminidase deficiency. The NAGA gene provides instructions for making the enzyme alpha-N-acetylgalactosaminidase. This enzyme works in the lysosomes, which are compartments within cells that digest and recycle materials. Alpha-N-acetylgalactosaminidase helps break down complexes called glycoproteins and glycolipids, which consist of sugar molecules attached to certain proteins and fats.
Variants in the NAGA gene can alter the alpha-N-acetylgalactosaminidase enzyme and interfere with the breakdown of glycoproteins and glycolipids. As these substances accumulate in the lysosomes, they can cause cells to malfunction and eventually die. Cell damage in the nervous system and other tissues and organs of the body leads to the signs and symptoms seen in people with alpha-N-acetylgalactosaminidase deficiency.
Researchers are studying why some people with NAGA gene variants have severe signs and symptoms, while others with the same gene variant have milder features or no features at all.
Conditions that cause molecules to build up inside lysosomes, such as alpha-N-acetylgalactosaminidase deficiency, belong to a large family of lysosomal storage disorders.
Inheritance
This condition is inherited in an autosomal recessive pattern , which means both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
, which means both copies of the gene in each cell must have a variant to cause the disorder. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
Other Names for This Condition
- Alpha-galactosidase B deficiency
- Alpha-galNAc deficiency, Schindler type
- Alpha-N-acetylgalactosaminidase deficiency
- Alpha-NAGA deficiency
- Angiokeratoma corporis diffusum-glycopeptiduria
- GALB deficiency
- Kanzaki disease
- Lysosomal glycoaminoacid storage disease-angiokeratoma corporis diffusum
- NAGA deficiency type I
- NAGA deficiency type II
- NAGA deficiency type III
- Neuroaxonal dystrophy, Schindler type
- Neuronal axonal dystrophy, Schindler type
- Schindler disease type I
- Schindler disease type II
- Schindler disease type III
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Bakker HD, de Sonnaville ML, Vreken P, Abeling NG, Groener JE, Keulemans JL, van Diggelen OP. Human alpha-N-acetylgalactosaminidase (alpha-NAGA) deficiency: no association with neuroaxonal dystrophy? Eur J Hum Genet. 2001 Feb;9(2):91-6. doi: 10.1038/sj.ejhg.5200598. Citation on PubMed
- Blanchon YC, Gay C, Gibert G, Lauras B. A case of N-acetyl galactosaminidase deficiency (Schindler disease) associated with autism. J Autism Dev Disord. 2002 Apr;32(2):145-6. doi: 10.1023/a:1017499407910. No abstract available. Citation on PubMed
- Castro RG, Perez AMG, Curto MCR, Alvarez JC, Ferreiros AC, Cuadros AV, Bueno DM, Fernandez AJC. A New Case of Schindler Disease. Eur J Case Rep Intern Med. 2019 Oct 25;6(11):001269. doi: 10.12890/2019_001269. eCollection 2019. Citation on PubMed
- Chabas A, Duque J, Gort L. A new infantile case of alpha-N-acetylgalactosaminidase deficiency. Cardiomyopathy as a presenting symptom. J Inherit Metab Dis. 2007 Feb;30(1):108. doi: 10.1007/s10545-006-0470-1. Epub 2006 Dec 14. Citation on PubMed
- Clark NE, Garman SC. The 1.9 a structure of human alpha-N-acetylgalactosaminidase: The molecular basis of Schindler and Kanzaki diseases. J Mol Biol. 2009 Oct 23;393(2):435-47. doi: 10.1016/j.jmb.2009.08.021. Epub 2009 Aug 14. Citation on PubMed or Free article on PubMed Central
- Desnick RJ, Wang AM. Schindler disease: an inherited neuroaxonal dystrophy due to alpha-N-acetylgalactosaminidase deficiency. J Inherit Metab Dis. 1990;13(4):549-59. doi: 10.1007/BF01799512. Citation on PubMed
- Kanda A, Tsuyama S, Murata F, Kodama K, Hirabayashi Y, Kanzaki T. Immunoelectron microscopic analysis of lysosomal deposits in alpha-N-acetylgalactosaminidase deficiency with angiokeratoma corporis diffusum. J Dermatol Sci. 2002 May;29(1):42-8. doi: 10.1016/s0923-1811(02)00005-1. Citation on PubMed
- Kanekura T, Sakuraba H, Matsuzawa F, Aikawa S, Doi H, Hirabayashi Y, Yoshii N, Fukushige T, Kanzaki T. Three dimensional structural studies of alpha-N-acetylgalactosaminidase (alpha-NAGA) in alpha-NAGA deficiency (Kanzaki disease): different gene mutations cause peculiar structural changes in alpha-NAGAs resulting in different substrate specificities and clinical phenotypes. J Dermatol Sci. 2005 Jan;37(1):15-20. doi: 10.1016/j.jdermsci.2004.09.005. Epub 2004 Dec 8. Citation on PubMed
- Kanzaki T, Yokota M, Irie F, Hirabayashi Y, Wang AM, Desnick RJ. Angiokeratoma corporis diffusum with glycopeptiduria due to deficient lysosomal alpha-N-acetylgalactosaminidase activity. Clinical, morphologic, and biochemical studies. Arch Dermatol. 1993 Apr;129(4):460-5. Citation on PubMed
- Kodama K, Kobayashi H, Abe R, Ohkawara A, Yoshii N, Yotsumoto S, Fukushige T, Nagatsuka Y, Hirabayashi Y, Kanzaki T. A new case of alpha-N-acetylgalactosaminidase deficiency with angiokeratoma corporis diffusum, with Meniere's syndrome and without mental retardation. Br J Dermatol. 2001 Feb;144(2):363-8. doi: 10.1046/j.1365-2133.2001.04028.x. Citation on PubMed
- Michalski JC, Klein A. Glycoprotein lysosomal storage disorders: alpha- and beta-mannosidosis, fucosidosis and alpha-N-acetylgalactosaminidase deficiency. Biochim Biophys Acta. 1999 Oct 8;1455(2-3):69-84. doi: 10.1016/s0925-4439(99)00077-0. Citation on PubMed
- Mohamed FE, Al Sorkhy M, Ghattas MA, Al-Zaabi N, Al-Shamsi A, Almansoori TM, Al-Gazali L, Al-Dirbashi OY, Al-Jasmi F, Ali BR. A Novel Homozygous Missense Variant in the NAGA Gene with Extreme Intrafamilial Phenotypic Heterogeneity. J Mol Neurosci. 2020 Jan;70(1):45-55. doi: 10.1007/s12031-019-01398-6. Epub 2019 Aug 29. Citation on PubMed
- Sakuraba H, Matsuzawa F, Aikawa SI, Doi H, Kotani M, Nakada H, Fukushige T, Kanzaki T. Structural and immunocytochemical studies on alpha-N-acetylgalactosaminidase deficiency (Schindler/Kanzaki disease). J Hum Genet. 2004;49(1):1-8. doi: 10.1007/s10038-003-0098-z. Epub 2003 Dec 19. Citation on PubMed
- Umehara F, Matsumuro K, Kurono Y, Arimura K, Osame M, Kanzaki T. Neurologic manifestations of Kanzaki disease. Neurology. 2004 May 11;62(9):1604-6. doi: 10.1212/01.wnl.0000123116.96441.34. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.