

Description
Allan-Herndon-Dudley syndrome is a rare disorder of brain development that causes moderate to severe intellectual disability and problems with movement. This condition, which occurs exclusively in males, disrupts development from before birth. Although affected males have impaired speech and a limited ability to communicate, they seem to enjoy interaction with other people.
Most children with Allan-Herndon-Dudley syndrome have weak muscle tone (hypotonia) and underdevelopment of many muscles (muscle hypoplasia). As they get older, they usually develop joint deformities called contractures, which restrict the movement of certain joints. Abnormal muscle stiffness (spasticity), muscle weakness, and involuntary movements of the arms and legs also limit mobility. As a result, many people with Allan-Herndon-Dudley syndrome are unable to walk independently and become wheelchair-bound by adulthood.
Frequency
Allan-Herndon-Dudley syndrome appears to be a rare disorder. About 25 families with individuals affected by this condition have been reported worldwide.
Causes
Mutations in the SLC16A2 gene cause Allan-Herndon-Dudley syndrome. The SLC16A2 gene, also known as MCT8, provides instructions for making a protein that plays a critical role in the development of the nervous system. This protein transports a particular hormone into nerve cells in the developing brain. This hormone, called triiodothyronine or T3, is produced by a butterfly-shaped gland in the lower neck called the thyroid. T3 appears to be critical for the normal formation and growth of nerve cells, as well as the development of junctions between nerve cells (synapses) where cell-to-cell communication occurs. T3 and other forms of thyroid hormone also help regulate the development of other organs and control the rate of chemical reactions in the body (metabolism).
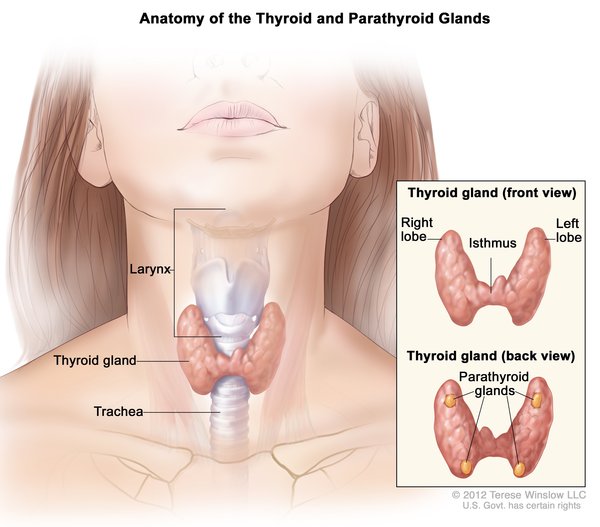


Gene mutations alter the structure and function of the SLC16A2 protein. As a result, this protein is unable to transport T3 into nerve cells effectively. A lack of this critical hormone in certain parts of the brain disrupts normal brain development, resulting in intellectual disability and problems with movement. Because T3 is not taken up by nerve cells, excess amounts of this hormone continue to circulate in the bloodstream. Increased T3 levels in the blood may be toxic to some organs and contribute to the signs and symptoms of Allan-Herndon-Dudley syndrome.
Inheritance
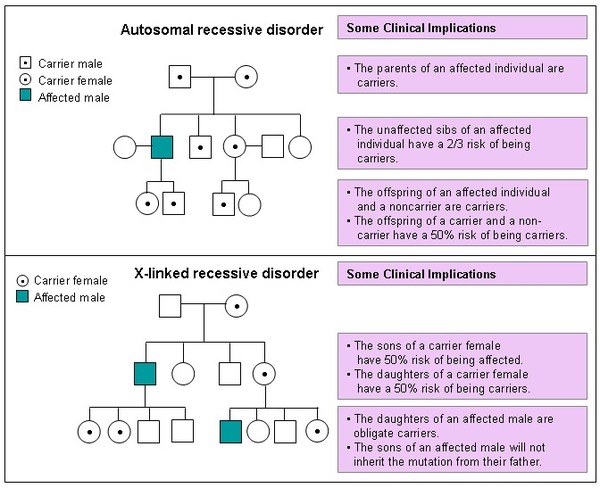
This condition is inherited in an X-linked recessive pattern. A condition is considered X-linked if the mutated gene that causes the disorder is located on the X chromosome, one of the two sex chromosomes. In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a mutation must be present in both copies of the gene to cause the disorder. Males are affected by X-linked recessive disorders much more frequently than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.

In X-linked recessive inheritance, a female with one altered copy of the gene in each cell is called a carrier. She can pass on the mutated gene, but usually does not experience signs and symptoms of the disorder. Carriers of SLC16A2 mutations have normal intelligence and do not experience problems with movement. Some carriers have been diagnosed with thyroid disease, a condition which is relatively common in the general population. It is unclear whether thyroid disease is related to SLC16A2 gene mutations in these cases.
Other Names for This Condition
- Allan-Herndon syndrome
- MCT8 (SLC16A2)-specific thyroid hormone cell transporter deficiency
- Mental retardation, X-linked, with hypotonia
- Monocarboxylate transporter 8 (MCT8) deficiency
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Dumitrescu AM, Liao XH, Weiss RE, Millen K, Refetoff S. Tissue-specific thyroid hormone deprivation and excess in monocarboxylate transporter (mct) 8-deficient mice. Endocrinology. 2006 Sep;147(9):4036-43. doi: 10.1210/en.2006-0390. Epub 2006 May 18. Citation on PubMed
- Friesema EC, Jansen J, Heuer H, Trajkovic M, Bauer K, Visser TJ. Mechanisms of disease: psychomotor retardation and high T3 levels caused by mutations in monocarboxylate transporter 8. Nat Clin Pract Endocrinol Metab. 2006 Sep;2(9):512-23. doi: 10.1038/ncpendmet0262. Citation on PubMed
- Herzovich V, Vaiani E, Marino R, Dratler G, Lazzati JM, Tilitzky S, Ramirez P, Iorcansky S, Rivarola MA, Belgorosky A. Unexpected peripheral markers of thyroid function in a patient with a novel mutation of the MCT8 thyroid hormone transporter gene. Horm Res. 2007;67(1):1-6. doi: 10.1159/000095805. Epub 2006 Sep 15. Citation on PubMed
- Holden KR, Zuniga OF, May MM, Su H, Molinero MR, Rogers RC, Schwartz CE. X-linked MCT8 gene mutations: characterization of the pediatric neurologic phenotype. J Child Neurol. 2005 Oct;20(10):852-7. doi: 10.1177/08830738050200101601. Citation on PubMed
- Jansen J, Friesema EC, Kester MH, Milici C, Reeser M, Gruters A, Barrett TG, Mancilla EE, Svensson J, Wemeau JL, Busi da Silva Canalli MH, Lundgren J, McEntagart ME, Hopper N, Arts WF, Visser TJ. Functional analysis of monocarboxylate transporter 8 mutations identified in patients with X-linked psychomotor retardation and elevated serum triiodothyronine. J Clin Endocrinol Metab. 2007 Jun;92(6):2378-81. doi: 10.1210/jc.2006-2570. Epub 2007 Mar 13. Citation on PubMed
- Kakinuma H, Itoh M, Takahashi H. A novel mutation in the monocarboxylate transporter 8 gene in a boy with putamen lesions and low free T4 levels in cerebrospinal fluid. J Pediatr. 2005 Oct;147(4):552-4. doi: 10.1016/j.jpeds.2005.05.012. Citation on PubMed
- Maranduba CM, Friesema EC, Kok F, Kester MH, Jansen J, Sertie AL, Passos-Bueno MR, Visser TJ. Decreased cellular uptake and metabolism in Allan-Herndon-Dudley syndrome (AHDS) due to a novel mutation in the MCT8 thyroid hormone transporter. J Med Genet. 2006 May;43(5):457-60. doi: 10.1136/jmg.2005.035840. Epub 2005 Jun 24. Citation on PubMed or Free article on PubMed Central
- Schwartz CE, May MM, Carpenter NJ, Rogers RC, Martin J, Bialer MG, Ward J, Sanabria J, Marsa S, Lewis JA, Echeverri R, Lubs HA, Voeller K, Simensen RJ, Stevenson RE. Allan-Herndon-Dudley syndrome and the monocarboxylate transporter 8 (MCT8) gene. Am J Hum Genet. 2005 Jul;77(1):41-53. doi: 10.1086/431313. Epub 2005 May 11. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.