

Description
ALG1-congenital disorder of glycosylation (ALG1-CDG, also known as congenital disorder of glycosylation type Ik) is an inherited disorder with varying signs and symptoms that typically develop during infancy and can affect several body systems.
Individuals with ALG1-CDG often have intellectual disability, delayed development, and weak muscle tone (hypotonia). Many affected individuals develop seizures that can be difficult to treat. Individuals with ALG1-CDG may also have movement problems such as involuntary rhythmic shaking (tremor) or difficulties with movement and balance (ataxia).
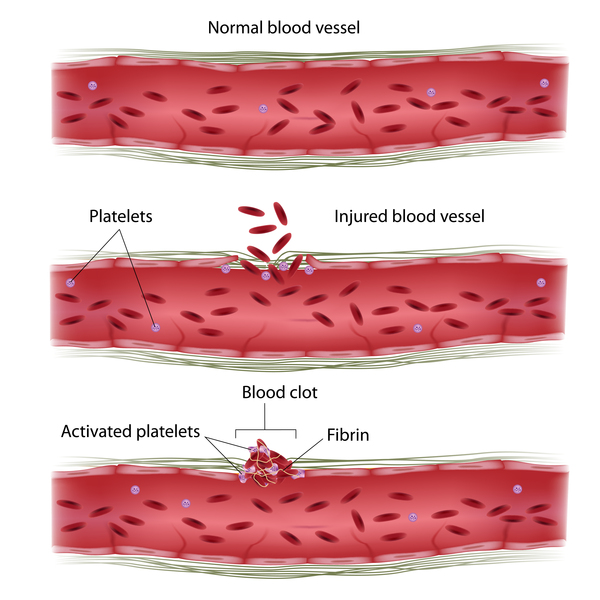
People with ALG1-CDG often have problems with blood clotting, which can lead to abnormal clotting or bleeding episodes. Additionally, affected individuals may produce abnormally low levels of proteins called antibodies (or immunoglobulins), particularly immunoglobulin G (IgG). Antibodies help protect the body against infection by foreign particles and germs. A reduction in antibodies can make it difficult for affected individuals to fight infections.

Some people with ALG1-CDG have physical abnormalities such as a small head size (microcephaly); unusual facial features; joint deformities called contractures; long, slender fingers and toes (arachnodactyly); or unusually fleshy pads at the tips of the fingers and toes. Eye problems that may occur in people with this condition include eyes that do not point in the same direction (strabismus) or involuntary eye movements (nystagmus). Rarely, affected individuals develop vision loss.

Less common abnormalities that occur in people with ALG1-CDG include respiratory problems, reduced sensation in their arms and legs (peripheral neuropathy), swelling (edema), and gastrointestinal difficulties.
The signs and symptoms of ALG1-CDG are often severe, with affected individuals surviving only into infancy or childhood. However, some people with this condition are more mildly affected and survive into adulthood.
Frequency
ALG1-CDG appears to be a rare disorder; fewer than 30 affected individuals have been described in the scientific literature.
Causes
Mutations in the ALG1 gene cause ALG1-CDG. This gene provides instructions for making an enzyme that is involved in a process called glycosylation. During this process, complex chains of sugar molecules (oligosaccharides) are added to proteins and fats (lipids). Glycosylation modifies proteins and lipids so they can fully perform their functions. The enzyme produced from the ALG1 gene transfers a simple sugar called mannose to growing oligosaccharides at a particular step in the formation of the sugar chain. Once the correct number of sugar molecules are linked together, the oligosaccharide is attached to a protein or lipid.
ALG1 gene mutations lead to the production of an abnormal enzyme with reduced activity. The poorly functioning enzyme cannot add mannose to sugar chains efficiently, and the resulting oligosaccharides are often incomplete. Although the short oligosaccharides can be transferred to proteins and fats, the process is not as efficient as with the full-length oligosaccharide. The wide variety of signs and symptoms in ALG1-CDG are likely due to impaired glycosylation of proteins and lipids that are needed for normal function of many organs and tissues.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- ALG1-CDG
- Carbohydrate deficient glycoprotein syndrome type Ik
- CDG1K
- CDGIk
- Congenital disorder of glycosylation type 1K
- Mannosyltransferase 1 deficiency
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Dupre T, Vuillaumier-Barrot S, Chantret I, Sadou Yaye H, Le Bizec C, Afenjar A, Altuzarra C, Barnerias C, Burglen L, de Lonlay P, Feillet F, Napuri S, Seta N, Moore SE. Guanosine diphosphate-mannose:GlcNAc2-PP-dolichol mannosyltransferase deficiency (congenital disorders of glycosylation type Ik): five new patients and seven novel mutations. J Med Genet. 2010 Nov;47(11):729-35. doi: 10.1136/jmg.2009.072504. Epub 2010 Aug 2. Citation on PubMed
- Jaeken J, Lefeber D, Matthijs G. Clinical utility gene card for: ALG1 defective congenital disorder of glycosylation. Eur J Hum Genet. 2015 Oct;23(10):1431. doi: 10.1038/ejhg.2015.9. Epub 2015 Feb 4. No abstract available. Citation on PubMed or Free article on PubMed Central
- Morava E, Vodopiutz J, Lefeber DJ, Janecke AR, Schmidt WM, Lechner S, Item CB, Sykut-Cegielska J, Adamowicz M, Wierzba J, Zhang ZH, Mihalek I, Stockler S, Bodamer OA, Lehle L, Wevers RA. Defining the phenotype in congenital disorder of glycosylation due to ALG1 mutations. Pediatrics. 2012 Oct;130(4):e1034-9. doi: 10.1542/peds.2011-2711. Epub 2012 Sep 10. Citation on PubMed
- Rohlfing AK, Rust S, Reunert J, Tirre M, Du Chesne I, Wemhoff S, Meinhardt F, Hartmann H, Das AM, Marquardt T. ALG1-CDG: a new case with early fatal outcome. Gene. 2014 Jan 25;534(2):345-51. doi: 10.1016/j.gene.2013.10.013. Epub 2013 Oct 21. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.