

Description
Adams-Oliver syndrome is a rare condition that is present at birth. The primary features are an abnormality in skin development (called aplasia cutis congenita) and malformations of the limbs. A variety of other features can occur in people with Adams-Oliver syndrome.
Most people with Adams-Oliver syndrome have aplasia cutis congenita, a condition characterized by localized areas of missing skin typically occurring on the top of the head (the skull vertex). In some cases, the bone under the skin is also underdeveloped. Individuals with this condition commonly have scarring and an absence of hair growth in the affected area.
Abnormalities of the hands and feet are also common in people with Adams-Oliver syndrome. These most often involve the fingers and toes and can include abnormal nails, fingers or toes that are fused together (syndactyly), and abnormally short or missing fingers or toes (brachydactyly or oligodactyly). In some cases, other bones in the hands, feet, or lower limbs are malformed or missing.
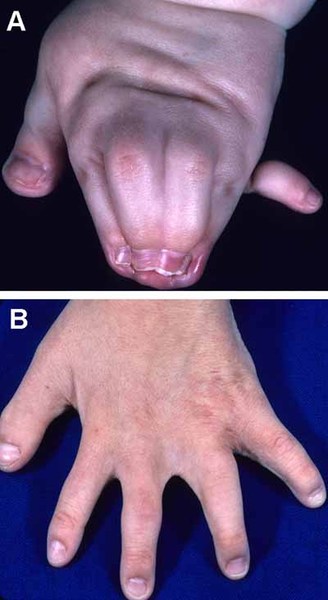
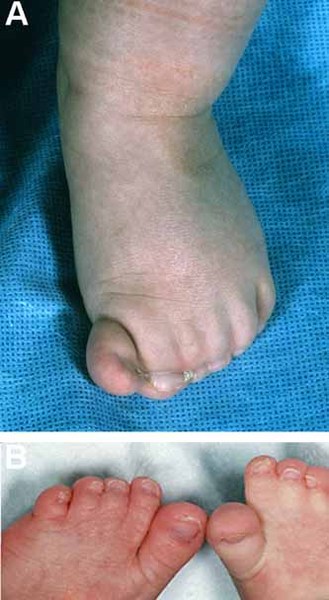
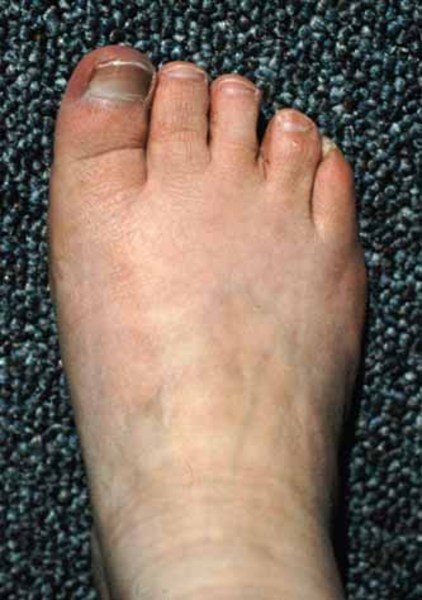

Some affected infants have a condition called cutis marmorata telangiectatica congenita. This disorder of the blood vessels causes a reddish or purplish net-like pattern on the skin. In addition, people with Adams-Oliver syndrome can develop high blood pressure in the blood vessels between the heart and the lungs (pulmonary hypertension), which can be life-threatening. Other blood vessel problems and heart defects can occur in affected individuals.
In some cases, people with Adams-Oliver syndrome have neurological problems, such as developmental delay, learning disabilities, or abnormalities in the structure of the brain.
Frequency
Adams-Oliver syndrome is a rare disorder; its prevalence is unknown.
Causes
Mutations in the ARHGAP31, DLL4, DOCK6, EOGT, NOTCH1, or RBPJ gene can cause Adams-Oliver syndrome. Because some affected individuals do not have mutations in one of these genes, it is likely that other genes that have not been identified are also involved in this condition. Each of the known genes plays an important role during embryonic development, and changes in any one of them can impair this tightly controlled process, leading to the signs and symptoms of Adams-Oliver syndrome.
The proteins produced from the ARHGAP31 and DOCK6 genes are both involved in the regulation of proteins called GTPases, which transmit signals that are critical for various aspects of embryonic development. The ARHGAP31 and DOCK6 proteins appear to be especially important for GTPase regulation during development of the limbs, skull, and heart. GTPases are often called molecular switches because they can be turned on and off. The DOCK6 protein turns them on, and the ARHGAP31 protein turns them off. Mutations in the DOCK6 gene lead to production of an abnormally short DOCK6 protein that is likely unable to turn on GTPases, which reduces their activity. Mutations in the ARHGAP31 gene also decrease GTPase activity by leading to production of an abnormally active ARHGAP31 protein, which turns off GTPases when it normally would not. This decline in GTPase activity leads to the skin problems, bone malformations, and other features characteristic of Adams-Oliver syndrome.
The proteins produced from the NOTCH1, DLL4, and RBPJ genes are part of a signaling pathway known as the Notch pathway. Notch signaling controls how certain types of cells develop in the growing embryo, including those that form the bones, heart, muscles, nerves, and blood vessels. The Notch1 and DLL4 proteins fit together like a lock and its key to stimulate one part of the Notch pathway, which is important for development of blood vessels. The NOTCH1 and DLL4 gene mutations involved in Adams-Oliver syndrome likely impair Notch1 signaling, which may underlie blood vessel and heart abnormalities in some people with Adams-Oliver syndrome. Researchers suspect that the other features of the condition may be due to abnormal blood vessel development before birth.
Signaling through Notch1 and other Notch proteins stimulates the RBP-J protein, produced from the RBPJ gene, to attach (bind) to specific regions of DNA and control the activity of genes that play a role in cellular development in multiple tissues throughout the body. The RBPJ gene mutations involved in Adams-Oliver syndrome alter the region of the RBP-J protein that normally binds DNA. The altered protein is unable to bind to DNA, preventing it from turning on particular genes. These changes in gene activity impair the proper development of the skin, bones, and other tissues, leading to the features of Adams-Oliver syndrome.
Little is known about how mutations in the EOGT gene cause Adams-Oliver syndrome. The protein produced from this gene modifies certain proteins by transferring a molecule called N-acetylglucosamine to them. It is thought that the EOGT protein modifies Notch proteins, which stimulate the Notch signaling pathway. However, the impact of the modification on Notch signaling is unclear. At least three mutations in the EOGT gene have been identified in people with Adams-Oliver syndrome, but how the genetic changes contribute to the signs and symptoms of this disorder is still unknown.
Inheritance
Adams-Oliver syndrome can have different inheritance patterns. When caused by mutations in the ARHGAP31, DLL4, NOTCH1, or RBPJ gene, the condition is inherited in an autosomal dominant pattern. Autosomal dominant inheritance means that one copy of the altered gene in each cell is sufficient to cause the disorder. The altered gene is typically inherited from an affected parent. Some cases associated with NOTCH1 gene mutations result from new (de novo) mutations in the gene that occur during the formation of reproductive cells (eggs or sperm) or in early embryonic development. These cases occur in people with no history of the disorder in their family.


When caused by mutations in the DOCK6 or EOGT gene, Adams-Oliver syndrome is inherited in an autosomal recessive pattern. In conditions with this pattern of inheritance, both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Absence defect of limbs, scalp, and skull
- AOS
- Aplasia cutis congenita with terminal transverse limb defects
- Congenital scalp defects with distal limb reduction anomalies
Additional Information & Resources
Genetic Testing Information
- Genetic Testing Registry: Adams-Oliver syndrome

- Genetic Testing Registry: Adams-Oliver syndrome 1
- Genetic Testing Registry: Adams-Oliver syndrome 2
- Genetic Testing Registry: Adams-Oliver syndrome 3
- Genetic Testing Registry: Adams-Oliver syndrome 4
- Genetic Testing Registry: Adams-Oliver syndrome 5
- Genetic Testing Registry: Adams-Oliver syndrome 6
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Cohen I, Silberstein E, Perez Y, Landau D, Elbedour K, Langer Y, Kadir R, Volodarsky M, Sivan S, Narkis G, Birk OS. Autosomal recessive Adams-Oliver syndrome caused by homozygous mutation in EOGT, encoding an EGF domain-specific O-GlcNAc transferase. Eur J Hum Genet. 2014 Mar;22(3):374-8. doi: 10.1038/ejhg.2013.159. Epub 2013 Jul 17. Citation on PubMed or Free article on PubMed Central
- Hassed SJ, Wiley GB, Wang S, Lee JY, Li S, Xu W, Zhao ZJ, Mulvihill JJ, Robertson J, Warner J, Gaffney PM. RBPJ mutations identified in two families affected by Adams-Oliver syndrome. Am J Hum Genet. 2012 Aug 10;91(2):391-5. doi: 10.1016/j.ajhg.2012.07.005. Citation on PubMed or Free article on PubMed Central
- Meester JA, Southgate L, Stittrich AB, Venselaar H, Beekmans SJ, den Hollander N, Bijlsma EK, Helderman-van den Enden A, Verheij JB, Glusman G, Roach JC, Lehman A, Patel MS, de Vries BB, Ruivenkamp C, Itin P, Prescott K, Clarke S, Trembath R, Zenker M, Sukalo M, Van Laer L, Loeys B, Wuyts W. Heterozygous Loss-of-Function Mutations in DLL4 Cause Adams-Oliver Syndrome. Am J Hum Genet. 2015 Sep 3;97(3):475-82. doi: 10.1016/j.ajhg.2015.07.015. Epub 2015 Aug 20. Citation on PubMed or Free article on PubMed Central
- Ogawa M, Sawaguchi S, Kawai T, Nadano D, Matsuda T, Yagi H, Kato K, Furukawa K, Okajima T. Impaired O-linked N-acetylglucosaminylation in the endoplasmic reticulum by mutated epidermal growth factor (EGF) domain-specific O-linked N-acetylglucosamine transferase found in Adams-Oliver syndrome. J Biol Chem. 2015 Jan 23;290(4):2137-49. doi: 10.1074/jbc.M114.598821. Epub 2014 Dec 8. Citation on PubMed or Free article on PubMed Central
- Shaheen R, Aglan M, Keppler-Noreuil K, Faqeih E, Ansari S, Horton K, Ashour A, Zaki MS, Al-Zahrani F, Cueto-Gonzalez AM, Abdel-Salam G, Temtamy S, Alkuraya FS. Mutations in EOGT confirm the genetic heterogeneity of autosomal-recessive Adams-Oliver syndrome. Am J Hum Genet. 2013 Apr 4;92(4):598-604. doi: 10.1016/j.ajhg.2013.02.012. Epub 2013 Mar 21. Citation on PubMed or Free article on PubMed Central
- Shaheen R, Faqeih E, Sunker A, Morsy H, Al-Sheddi T, Shamseldin HE, Adly N, Hashem M, Alkuraya FS. Recessive mutations in DOCK6, encoding the guanidine nucleotide exchange factor DOCK6, lead to abnormal actin cytoskeleton organization and Adams-Oliver syndrome. Am J Hum Genet. 2011 Aug 12;89(2):328-33. doi: 10.1016/j.ajhg.2011.07.009. Epub 2011 Aug 4. Citation on PubMed or Free article on PubMed Central
- Snape KM, Ruddy D, Zenker M, Wuyts W, Whiteford M, Johnson D, Lam W, Trembath RC. The spectra of clinical phenotypes in aplasia cutis congenita and terminal transverse limb defects. Am J Med Genet A. 2009 Aug;149A(8):1860-81. doi: 10.1002/ajmg.a.32708. Citation on PubMed
- Southgate L, Machado RD, Snape KM, Primeau M, Dafou D, Ruddy DM, Branney PA, Fisher M, Lee GJ, Simpson MA, He Y, Bradshaw TY, Blaumeiser B, Winship WS, Reardon W, Maher ER, FitzPatrick DR, Wuyts W, Zenker M, Lamarche-Vane N, Trembath RC. Gain-of-function mutations of ARHGAP31, a Cdc42/Rac1 GTPase regulator, cause syndromic cutis aplasia and limb anomalies. Am J Hum Genet. 2011 May 13;88(5):574-85. doi: 10.1016/j.ajhg.2011.04.013. Citation on PubMed or Free article on PubMed Central
- Southgate L, Sukalo M, Karountzos ASV, Taylor EJ, Collinson CS, Ruddy D, Snape KM, Dallapiccola B, Tolmie JL, Joss S, Brancati F, Digilio MC, Graul-Neumann LM, Salviati L, Coerdt W, Jacquemin E, Wuyts W, Zenker M, Machado RD, Trembath RC. Haploinsufficiency of the NOTCH1 Receptor as a Cause of Adams-Oliver Syndrome With Variable Cardiac Anomalies. Circ Cardiovasc Genet. 2015 Aug;8(4):572-581. doi: 10.1161/CIRCGENETICS.115.001086. Epub 2015 May 11. Citation on PubMed or Free article on PubMed Central
- Stittrich AB, Lehman A, Bodian DL, Ashworth J, Zong Z, Li H, Lam P, Khromykh A, Iyer RK, Vockley JG, Baveja R, Silva ES, Dixon J, Leon EL, Solomon BD, Glusman G, Niederhuber JE, Roach JC, Patel MS. Mutations in NOTCH1 cause Adams-Oliver syndrome. Am J Hum Genet. 2014 Sep 4;95(3):275-84. doi: 10.1016/j.ajhg.2014.07.011. Epub 2014 Aug 14. Citation on PubMed or Free article on PubMed Central
- Sukalo M, Tilsen F, Kayserili H, Muller D, Tuysuz B, Ruddy DM, Wakeling E, Orstavik KH, Snape KM, Trembath R, De Smedt M, van der Aa N, Skalej M, Mundlos S, Wuyts W, Southgate L, Zenker M. DOCK6 mutations are responsible for a distinct autosomal-recessive variant of Adams-Oliver syndrome associated with brain and eye anomalies. Hum Mutat. 2015 Jun;36(6):593-8. doi: 10.1002/humu.22795. Epub 2015 Apr 21. Citation on PubMed
- Swartz EN, Sanatani S, Sandor GG, Schreiber RA. Vascular abnormalities in Adams-Oliver syndrome: cause or effect? Am J Med Genet. 1999 Jan 1;82(1):49-52. doi: 10.1002/(sici)1096-8628(19990101)82:13.0.co;2-m. Citation on PubMed
- Verdyck P, Holder-Espinasse M, Hul WV, Wuyts W. Clinical and molecular analysis of nine families with Adams-Oliver syndrome. Eur J Hum Genet. 2003 Jun;11(6):457-63. doi: 10.1038/sj.ejhg.5200980. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.