

Description
Actin-accumulation myopathy is a disorder that primarily affects skeletal muscles, which are muscles that the body uses for movement. People with actin-accumulation myopathy have severe muscle weakness (myopathy) and poor muscle tone (hypotonia) throughout the body. Signs and symptoms of this condition are apparent in infancy and include feeding and swallowing difficulties, a weak cry, and difficulty with controlling head movements. Affected babies are sometimes described as "floppy" and may be unable to move on their own.
The severe muscle weakness that occurs in actin-accumulation myopathy also affects the muscles used for breathing. Individuals with this disorder may take shallow breaths (hypoventilate), especially during sleep, resulting in a shortage of oxygen and a buildup of carbon dioxide in the blood. Frequent respiratory infections and life-threatening breathing difficulties can occur. Because of the respiratory problems, most affected individuals do not survive past infancy. Those who do survive have delayed development of motor skills such as sitting, crawling, standing, and walking.
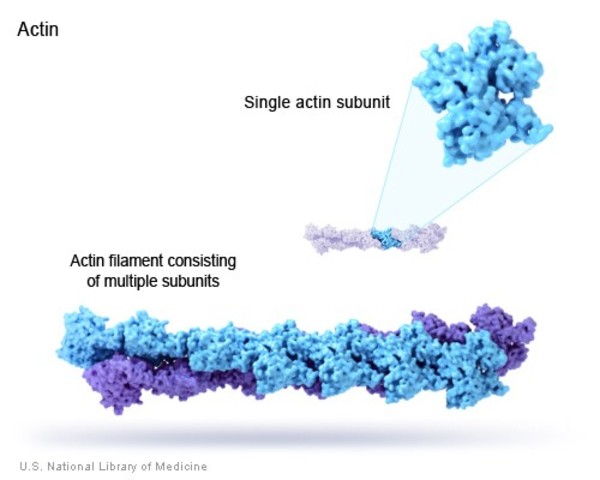
The name actin-accumulation myopathy derives from characteristic accumulations in muscle cells of filaments composed of a protein called actin. These filaments can be seen when muscle tissue is viewed under a microscope.
Frequency
Actin-accumulation myopathy is a rare disorder that has been identified in only a small number of individuals. Its exact prevalence is unknown.
Causes
Actin-accumulation myopathy is caused by a mutation in the ACTA1 gene. This gene provides instructions for making a protein called skeletal alpha (α)-actin, which is a member of the actin protein family found in skeletal muscles. Actin proteins are important for cell movement and the tensing of muscle fibers (muscle contraction). Thin filaments made up of actin molecules and thick filaments made up of another protein called myosin are the primary components of muscle fibers and are important for muscle contraction. Attachment (binding) and release of the overlapping thick and thin filaments allows them to move relative to each other so that the muscles can contract.
ACTA1 gene mutations that cause actin-accumulation myopathy may affect the way the skeletal α-actin protein binds to ATP. ATP is a molecule that supplies energy for cells' activities, and is important in the formation of thin filaments from individual actin molecules. Dysfunctional actin-ATP binding may result in abnormal thin filament formation and impair muscle contraction, leading to muscle weakness and the other signs and symptoms of actin-accumulation myopathy.
In some people with actin-accumulation myopathy, no ACTA1 gene mutations have been identified. The cause of the disorder in these individuals is unknown.
Inheritance
Actin-accumulation myopathy is an autosomal dominant condition, which means one copy of the altered gene in each cell is sufficient to cause the disorder. Most cases are not inherited; they result from new (de novo) mutations in the gene and occur in people with no history of the disorder in their family.

Other Names for This Condition
- Actin filament aggregate myopathy
- Actin myopathy
- Congenital myopathy with excess of thin filaments
- Nemaline myopathy 3
Additional Information & Resources
Genetic Testing Information
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Bornemann A, Petersen MB, Schmalbruch H. Fatal congenital myopathy with actin filament deposits. Acta Neuropathol. 1996 Jul;92(1):104-8. doi: 10.1007/s004010050496. Citation on PubMed
- Feng JJ, Marston S. Genotype-phenotype correlations in ACTA1 mutations that cause congenital myopathies. Neuromuscul Disord. 2009 Jan;19(1):6-16. doi: 10.1016/j.nmd.2008.09.005. Epub 2008 Oct 30. Citation on PubMed
- Goebel HH, Anderson JR, Hubner C, Oexle K, Warlo I. Congenital myopathy with excess of thin myofilaments. Neuromuscul Disord. 1997 May;7(3):160-8. doi: 10.1016/s0960-8966(97)00441-0. Citation on PubMed
- Laing NG, Dye DE, Wallgren-Pettersson C, Richard G, Monnier N, Lillis S, Winder TL, Lochmuller H, Graziano C, Mitrani-Rosenbaum S, Twomey D, Sparrow JC, Beggs AH, Nowak KJ. Mutations and polymorphisms of the skeletal muscle alpha-actin gene (ACTA1). Hum Mutat. 2009 Sep;30(9):1267-77. doi: 10.1002/humu.21059. Citation on PubMed or Free article on PubMed Central
- Nowak KJ, Ravenscroft G, Laing NG. Skeletal muscle alpha-actin diseases (actinopathies): pathology and mechanisms. Acta Neuropathol. 2013 Jan;125(1):19-32. doi: 10.1007/s00401-012-1019-z. Epub 2012 Jul 24. Citation on PubMed
- Nowak KJ, Wattanasirichaigoon D, Goebel HH, Wilce M, Pelin K, Donner K, Jacob RL, Hubner C, Oexle K, Anderson JR, Verity CM, North KN, Iannaccone ST, Muller CR, Nurnberg P, Muntoni F, Sewry C, Hughes I, Sutphen R, Lacson AG, Swoboda KJ, Vigneron J, Wallgren-Pettersson C, Beggs AH, Laing NG. Mutations in the skeletal muscle alpha-actin gene in patients with actin myopathy and nemaline myopathy. Nat Genet. 1999 Oct;23(2):208-12. doi: 10.1038/13837. Citation on PubMed
- Ochala J. Thin filament proteins mutations associated with skeletal myopathies: defective regulation of muscle contraction. J Mol Med (Berl). 2008 Nov;86(11):1197-204. doi: 10.1007/s00109-008-0380-9. Epub 2008 Jun 24. Citation on PubMed
- Schroder JM, Durling H, Laing N. Actin myopathy with nemaline bodies, intranuclear rods, and a heterozygous mutation in ACTA1 (Asp154Asn). Acta Neuropathol. 2004 Sep;108(3):250-6. doi: 10.1007/s00401-004-0888-1. Epub 2004 Jun 24. Citation on PubMed
- Sparrow JC, Nowak KJ, Durling HJ, Beggs AH, Wallgren-Pettersson C, Romero N, Nonaka I, Laing NG. Muscle disease caused by mutations in the skeletal muscle alpha-actin gene (ACTA1). Neuromuscul Disord. 2003 Sep;13(7-8):519-31. doi: 10.1016/s0960-8966(03)00101-9. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.