

Description
Achondrogenesis is a group of severe disorders that affect cartilage and bone development. These conditions are characterized by skeletal abnormalities that cause serious health problems. As a result, most infants with achondrogenesis die before birth or soon after, often due to respiratory failure.
Researchers have described three main types of achondrogenesis: type 1A, type 1B, and type 2. While these types differ in their genetic causes and inheritance patterns, they often have overlapping signs and symptoms. Genetic testing and medical imaging are often needed to tell them apart.
All forms of achondrogenesis feature short arms and legs, a narrow chest, and underdeveloped lungs. Infants with achondrogenesis type 1A, which is also called TRIP11-related achondrogenesis, typically have ribs that fracture easily. Bone formation (ossification) is also severely reduced in the skull and spine.
Infants with achondrogenesis type 1B, which is also called SLC26A2-related achondrogenesis, often have short fingers and toes and feet that may turn inward and upward (clubfeet ). Infants with achondrogenesis type 1B may also have a sac (pouch) formed from the inner lining of the abdominal cavity that pushes through a hole in the abdominal wall around the belly-button (umbilical hernia
). Infants with achondrogenesis type 1B may also have a sac (pouch) formed from the inner lining of the abdominal cavity that pushes through a hole in the abdominal wall around the belly-button (umbilical hernia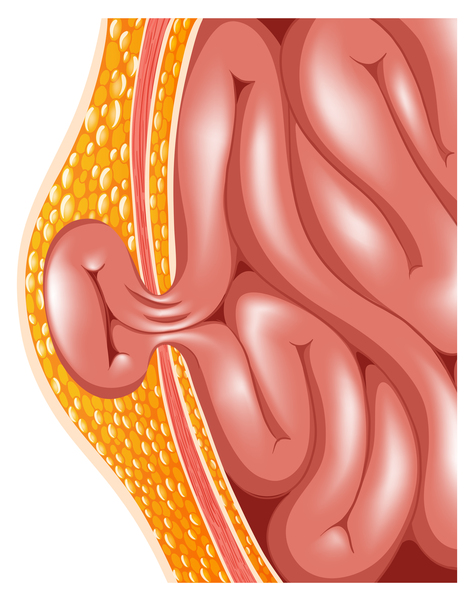 ) or near the groin (inguinal hernia).
) or near the groin (inguinal hernia).
The ossification of the spine and pelvis may be severely reduced in infants with achondrogenesis type 2, which is also called COL2A1-related achondrogenesis. The distinctive facial features seen in infants with achondrogenesis type 2 include a prominent forehead, a small chin, and, in some cases, an opening in the roof of the mouth (cleft palate ).
).
Achondrogenesis type 2 and a similar skeletal disorder called hypochondrogenesis were once thought to be distinct conditions. However, because these conditions have overlapping features and a shared genetic cause, they are now considered to be part of the same disease spectrum.
Frequency
Achondrogenesis occurs in approximately 1 in 40,000 to 60,000 newborns.
Causes
Variants (also called mutations) in different genes cause the three main types of achondrogenesis.
Variants in the TRIP11 gene cause achondrogenesis type 1A. This gene provides instructions for making a protein called thyroid receptor-interacting protein 11 (TRIP-11). TRIP-11 helps maintain the Golgi apparatus, a cell structure in which proteins are modified. Most of the variants in the TRIP11 gene that cause achondrogenesis type 1A prevent the production of functional TRIP-11 proteins. Researchers suspect that cells called chondrocytes may be sensitive to these changes. Chondrocytes give rise to cartilage, a tough, flexible tissue that makes up much of the skeleton during early development. Without enough functional TRIP-11, the Golgi apparatus cannot work properly, which likely contributes to the characteristic features of achondrogenesis type 1A.
Achondrogenesis type 1B is caused by variants in the SLC26A2 gene. This gene provides instructions for making a protein that transports charged molecules (ions), particularly sulfate ions, across cell membranes. This protein is essential for the normal development of cartilage and for converting cartilage into bone. Variants in the SLC26A2 gene disrupt the structure of developing cartilage, which prevents bones from forming properly. These changes cause the skeletal problems that are characteristic of achondrogenesis type 1B.
Achondrogenesis type 2 and hypochondrogenesis are caused by variants in the COL2A1 gene. This gene provides instructions for making a protein that forms type II collagen, which is found in the clear gel that fills the eyeball (the vitreous) and in cartilage. Type II collagen is essential for the normal growth and development of bones and other connective tissues . Variants in the COL2A1 gene interfere with the formation of mature type II collagen molecules, which prevents bones and other connective tissues from developing properly in people with achondrogenesis type 2 and hypochondrogenesis.
. Variants in the COL2A1 gene interfere with the formation of mature type II collagen molecules, which prevents bones and other connective tissues from developing properly in people with achondrogenesis type 2 and hypochondrogenesis.
Inheritance
Achondrogenesis type 1A and type 1B both have an autosomal recessive pattern of inheritance. A variant must be present in both copies of the TRIP11 gene in each cell to cause achondrogenesis type 1A, while a variant must be present in both copies of the SLC26A2 gene in each cell to cause achondrogenesis type 1B. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
of inheritance. A variant must be present in both copies of the TRIP11 gene in each cell to cause achondrogenesis type 1A, while a variant must be present in both copies of the SLC26A2 gene in each cell to cause achondrogenesis type 1B. The parents of an individual with an autosomal recessive condition each carry one copy of the altered gene, but they typically do not show signs and symptoms of the condition.
Achondrogenesis type 2 is considered to be an autosomal dominant disorder because one copy of the altered gene in each cell is sufficient to cause the condition. Most cases of this condition are the result of a new (de novo) variant in the COL2A1 gene that occurs during the formation of reproductive cells (eggs or sperm) in an affected individual's parent or during early embryonic development. These affected individuals have no history of the disorder in their family.
disorder because one copy of the altered gene in each cell is sufficient to cause the condition. Most cases of this condition are the result of a new (de novo) variant in the COL2A1 gene that occurs during the formation of reproductive cells (eggs or sperm) in an affected individual's parent or during early embryonic development. These affected individuals have no history of the disorder in their family.
Other Names for This Condition
- achondrogenesis type IA (Houston-Harris type)
- achondrogenesis type IB (Fraccaro type)
- achondrogenesis type II (Langer-Saldino type)
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Barat-Houari M, Sarrabay G, Gatinois V, Fabre A, Dumont B, Genevieve D, Touitou I. Mutation Update for COL2A1 Gene Variants Associated with Type II Collagenopathies. Hum Mutat. 2016 Jan;37(1):7-15. doi: 10.1002/humu.22915. Epub 2015 Oct 21. Citation on PubMed
- Borochowitz Z, Ornoy A, Lachman R, Rimoin DL. Achondrogenesis II-hypochondrogenesis: variability versus heterogeneity. Am J Med Genet. 1986 Jun;24(2):273-88. doi: 10.1002/ajmg.1320240208. Citation on PubMed
- Faivre L, Le Merrer M, Douvier S, Laurent N, Thauvin-Robinet C, Rousseau T, Vereecke I, Sagot P, Delezoide AL, Coucke P, Mortier G. Recurrence of achondrogenesis type II within the same family: evidence for germline mosaicism. Am J Med Genet A. 2004 Apr 30;126A(3):308-12. doi: 10.1002/ajmg.a.20597. Citation on PubMed
- Gregersen PA, Savarirayan R. Type II Collagen Disorders Overview. 2019 Apr 25 [updated 2024 Oct 24]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK540447/ Citation on PubMed
- Grigelioniene G, Geiberger S, Papadogiannakis N, Makitie O, Nishimura G, Nordgren A, Conner P. The phenotype range of achondrogenesis 1A. Am J Med Genet A. 2013 Oct;161A(10):2554-8. doi: 10.1002/ajmg.a.36106. Epub 2013 Aug 16. Citation on PubMed
- Kapur RP. Achondrogenesis. Pediatr Dev Pathol. 2007 Jul-Aug;10(4):253-5. doi: 10.2350/07-01-0216.1. Citation on PubMed
- Korkko J, Cohn DH, Ala-Kokko L, Krakow D, Prockop DJ. Widely distributed mutations in the COL2A1 gene produce achondrogenesis type II/hypochondrogenesis. Am J Med Genet. 2000 May 15;92(2):95-100. doi: 10.1002/(sici)1096-8628(20000515)92:23.0.co;2-9. Citation on PubMed
- Rossi A, Superti-Furga A. Mutations in the diastrophic dysplasia sulfate transporter (DTDST) gene (SLC26A2): 22 novel mutations, mutation review, associated skeletal phenotypes, and diagnostic relevance. Hum Mutat. 2001 Mar;17(3):159-71. doi: 10.1002/humu.1. Citation on PubMed
- Smits P, Bolton AD, Funari V, Hong M, Boyden ED, Lu L, Manning DK, Dwyer ND, Moran JL, Prysak M, Merriman B, Nelson SF, Bonafe L, Superti-Furga A, Ikegawa S, Krakow D, Cohn DH, Kirchhausen T, Warman ML, Beier DR. Lethal skeletal dysplasia in mice and humans lacking the golgin GMAP-210. N Engl J Med. 2010 Jan 21;362(3):206-16. doi: 10.1056/NEJMoa0900158. Citation on PubMed or Free article on PubMed Central
- Superti-Furga A, Hastbacka J, Wilcox WR, Cohn DH, van der Harten HJ, Rossi A, Blau N, Rimoin DL, Steinmann B, Lander ES, Gitzelmann R. Achondrogenesis type IB is caused by mutations in the diastrophic dysplasia sulphate transporter gene. Nat Genet. 1996 Jan;12(1):100-2. doi: 10.1038/ng0196-100. No abstract available. Citation on PubMed
- Unger S, Ferreira CR, Mortier GR, Ali H, Bertola DR, Calder A, Cohn DH, Cormier-Daire V, Girisha KM, Hall C, Krakow D, Makitie O, Mundlos S, Nishimura G, Robertson SP, Savarirayan R, Sillence D, Simon M, Sutton VR, Warman ML, Superti-Furga A. Nosology of genetic skeletal disorders: 2023 revision. Am J Med Genet A. 2023 May;191(5):1164-1209. doi: 10.1002/ajmg.a.63132. Epub 2023 Feb 13. Citation on PubMed
- Unger S, Superti-Furga A. Achondrogenesis Type 1B. 2002 Aug 30 [updated 2023 Mar 16]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK1516/ Citation on PubMed
- Vanegas S, Sua LF, Lopez-Tenorio J, Ramirez-Montano D, Pachajoa H. Achondrogenesis type 1A: clinical, histologic, molecular, and prenatal ultrasound diagnosis. Appl Clin Genet. 2018 May 25;11:69-73. doi: 10.2147/TACG.S157235. eCollection 2018. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.