

Description
ACAD9 deficiency is a condition that varies in severity and can cause muscle weakness (myopathy), heart problems, and intellectual disability. Nearly all affected individuals have a buildup of a chemical called lactic acid in the body (lactic acidosis). Additional signs and symptoms that affect other body systems occur in rare cases.
Mildly affected individuals with ACAD9 deficiency usually experience nausea and extreme fatigue in response to physical activity (exercise intolerance). People with ACAD9 deficiency who are moderately affected have low muscle tone (hypotonia) and weakness in the muscles used for movement (skeletal muscles). Severely affected individuals have brain dysfunction combined with myopathy (encephalomyopathy); these individuals usually also have an enlarged and weakened heart muscle (hypertrophic cardiomyopathy), which is typically fatal in infancy or childhood.
Individuals with ACAD9 deficiency who survive past early childhood often have intellectual disability and may develop seizures. Rare signs and symptoms of ACAD9 deficiency include movement disorders and problems with liver and kidney function.
Some individuals with ACAD9 deficiency have had improvement in muscle strength and a reduction in lactic acid levels with treatment.
Frequency
The prevalence of ACAD9 deficiency is unknown. At least 25 people with this condition have been described in the scientific literature.
Causes
ACAD9 deficiency is caused by mutations in the ACAD9 gene. This gene provides instructions for making an enzyme that is critical in helping assemble a group of proteins known as complex I. Complex I is found in mitochondria, which are the energy-producing structures inside cells. Complex I is one of several complexes that carry out a multistep process called oxidative phosphorylation, through which cells derive much of their energy.

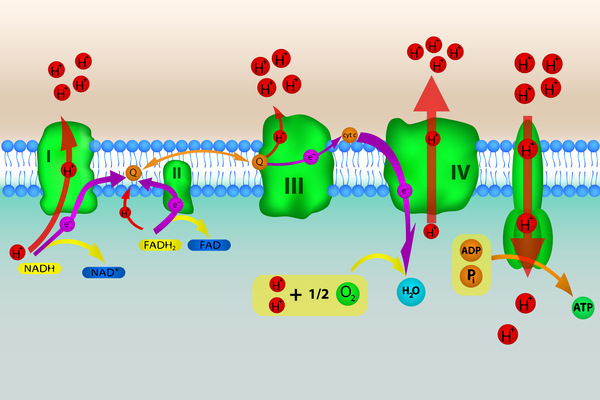
The ACAD9 enzyme also plays a role in fatty acid oxidation, a multistep process that occurs within mitochondria to break down (metabolize) fats and convert them into energy. The ACAD9 enzyme helps metabolize a certain group of fats called long-chain fatty acids. Fatty acids are a major source of energy for the heart and muscles. During periods without food (fasting), fatty acids are also an important energy source for the liver and other tissues.
Some ACAD9 gene mutations disrupt complex I assembly as well as long-chain fatty acid oxidation, while others affect only complex I assembly. The mutations that affect both of the enzyme's functions tend to be associated with the most severe signs and symptoms of ACAD9 deficiency, such as encephalomyopathy and hypertrophic cardiomyopathy. Although the exact mechanism is unclear, it is likely that cells that are less able to produce energy die off, particularly cells in the brain, skeletal muscle, and other tissues and organs that require a lot of energy. The loss of cells in these tissues is thought to lead to the signs and symptoms of ACAD9 deficiency.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- Acyl-CoA dehydrogenase 9 deficiency
- Deficiency of acyl-CoA dehydrogenase family member 9
- Mitochondrial complex I deficiency due to ACAD9 deficiency
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Aintablian HK, Narayanan V, Belnap N, Ramsey K, Grebe TA. An atypical presentation of ACAD9 deficiency: Diagnosis by whole exome sequencing broadens the phenotypic spectrum and alters treatment approach. Mol Genet Metab Rep. 2016 Dec 29;10:38-44. doi: 10.1016/j.ymgmr.2016.12.005. eCollection 2017 Mar. Citation on PubMed or Free article on PubMed Central
- Collet M, Assouline Z, Bonnet D, Rio M, Iserin F, Sidi D, Goldenberg A, Lardennois C, Metodiev MD, Haberberger B, Haack T, Munnich A, Prokisch H, Rotig A. High incidence and variable clinical outcome of cardiac hypertrophy due to ACAD9 mutations in childhood. Eur J Hum Genet. 2016 Aug;24(8):1112-6. doi: 10.1038/ejhg.2015.264. Epub 2015 Dec 16. Citation on PubMed or Free article on PubMed Central
- Dewulf JP, Barrea C, Vincent MF, De Laet C, Van Coster R, Seneca S, Marie S, Nassogne MC. Evidence of a wide spectrum of cardiac involvement due to ACAD9 mutations: Report on nine patients. Mol Genet Metab. 2016 Jul;118(3):185-189. doi: 10.1016/j.ymgme.2016.05.005. Epub 2016 May 13. Citation on PubMed
- Gerards M, van den Bosch BJ, Danhauser K, Serre V, van Weeghel M, Wanders RJ, Nicolaes GA, Sluiter W, Schoonderwoerd K, Scholte HR, Prokisch H, Rotig A, de Coo IF, Smeets HJ. Riboflavin-responsive oxidative phosphorylation complex I deficiency caused by defective ACAD9: new function for an old gene. Brain. 2011 Jan;134(Pt 1):210-9. doi: 10.1093/brain/awq273. Epub 2010 Oct 7. Citation on PubMed
- Haack TB, Danhauser K, Haberberger B, Hoser J, Strecker V, Boehm D, Uziel G, Lamantea E, Invernizzi F, Poulton J, Rolinski B, Iuso A, Biskup S, Schmidt T, Mewes HW, Wittig I, Meitinger T, Zeviani M, Prokisch H. Exome sequencing identifies ACAD9 mutations as a cause of complex I deficiency. Nat Genet. 2010 Dec;42(12):1131-4. doi: 10.1038/ng.706. Epub 2010 Nov 7. Citation on PubMed
- Nouws J, Nijtmans L, Houten SM, van den Brand M, Huynen M, Venselaar H, Hoefs S, Gloerich J, Kronick J, Hutchin T, Willems P, Rodenburg R, Wanders R, van den Heuvel L, Smeitink J, Vogel RO. Acyl-CoA dehydrogenase 9 is required for the biogenesis of oxidative phosphorylation complex I. Cell Metab. 2010 Sep 8;12(3):283-94. doi: 10.1016/j.cmet.2010.08.002. Citation on PubMed
- Nouws J, Te Brinke H, Nijtmans LG, Houten SM. ACAD9, a complex I assembly factor with a moonlighting function in fatty acid oxidation deficiencies. Hum Mol Genet. 2014 Mar 1;23(5):1311-9. doi: 10.1093/hmg/ddt521. Epub 2013 Oct 24. Citation on PubMed
- Schiff M, Haberberger B, Xia C, Mohsen AW, Goetzman ES, Wang Y, Uppala R, Zhang Y, Karunanidhi A, Prabhu D, Alharbi H, Prochownik EV, Haack T, Haberle J, Munnich A, Rotig A, Taylor RW, Nicholls RD, Kim JJ, Prokisch H, Vockley J. Complex I assembly function and fatty acid oxidation enzyme activity of ACAD9 both contribute to disease severity in ACAD9 deficiency. Hum Mol Genet. 2015 Jun 1;24(11):3238-47. doi: 10.1093/hmg/ddv074. Epub 2015 Feb 26. Citation on PubMed or Free article on PubMed Central
- Schrank B, Schoser B, Klopstock T, Schneiderat P, Horvath R, Abicht A, Holinski-Feder E, Augustis S. Lifetime exercise intolerance with lactic acidosis as key manifestation of novel compound heterozygous ACAD9 mutations causing complex I deficiency. Neuromuscul Disord. 2017 May;27(5):473-476. doi: 10.1016/j.nmd.2017.02.005. Epub 2017 Feb 14. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.