

Description
3-M syndrome is a disorder that causes skeletal abnormalities including short stature (dwarfism) and unusual facial features. The name of this condition comes from the initials of three researchers who first identified it: Miller, McKusick, and Malvaux.
Individuals with 3-M syndrome grow extremely slowly before birth, and this slow growth continues throughout childhood and adolescence. They have low birth weight and length and remain much smaller than others in their family, growing to an adult height of approximately 4 feet to 4 feet 6 inches (120 centimeters to 130 centimeters). In some affected individuals, the head is normal-sized but looks disproportionately large in comparison with the body. In other people with this disorder, the head has an unusually long and narrow shape (dolichocephaly). Intelligence is unaffected by 3-M syndrome, and life expectancy is generally normal.
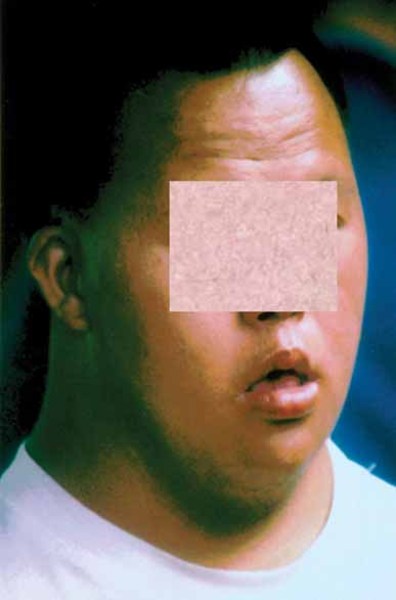
In addition to short stature, people with 3-M syndrome have a triangle-shaped face with a broad, prominent forehead (frontal bossing) and a pointed chin; the middle of the face is less prominent (hypoplastic midface). Other common features include large ears, full eyebrows, an upturned nose with a fleshy tip, a long area between the nose and mouth (philtrum), a prominent mouth, and full lips.
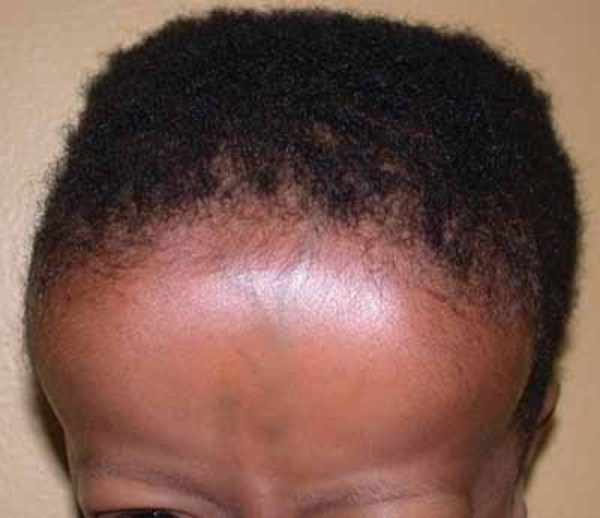
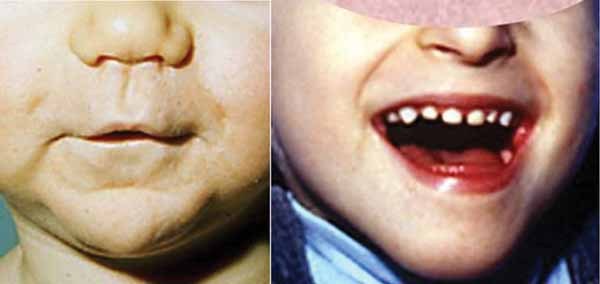
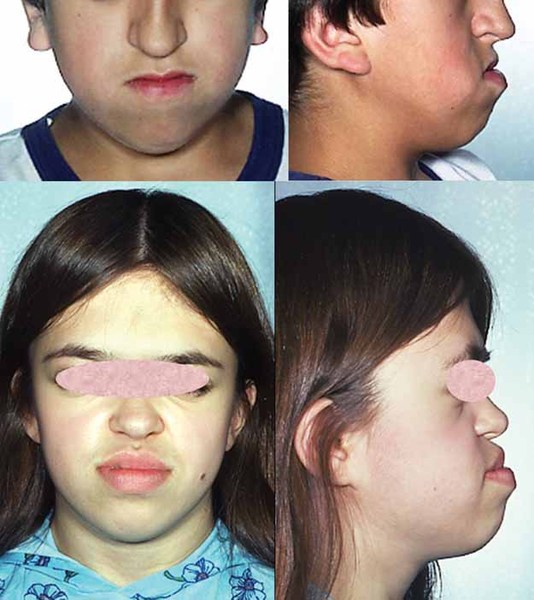
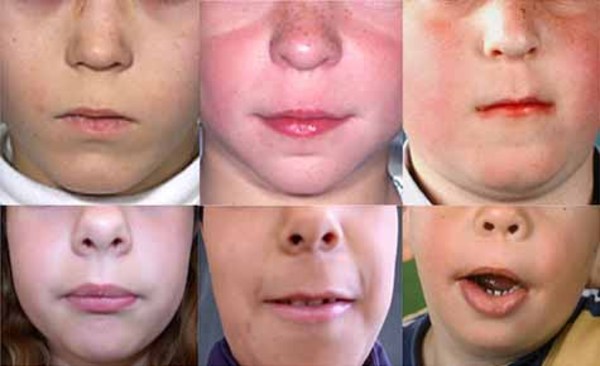
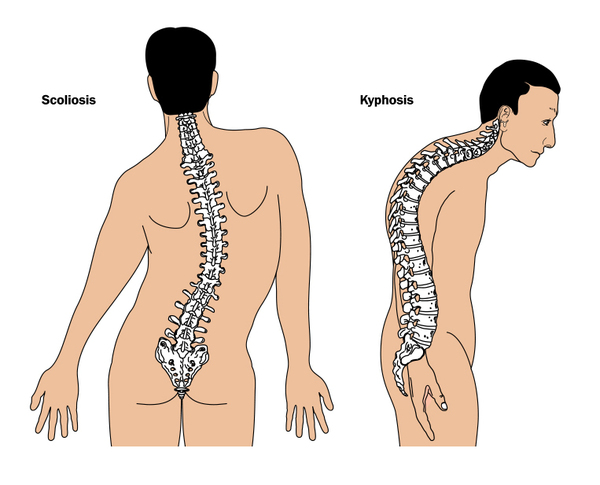
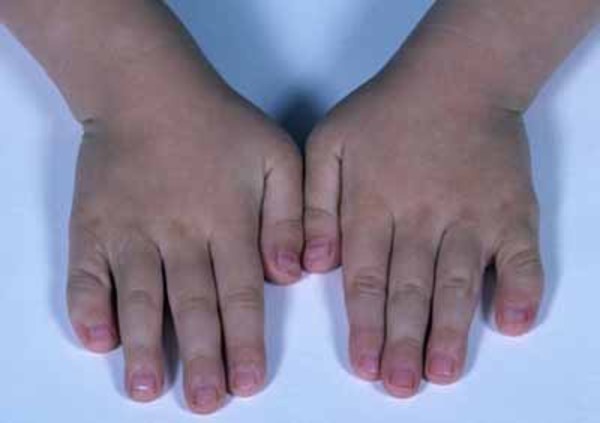
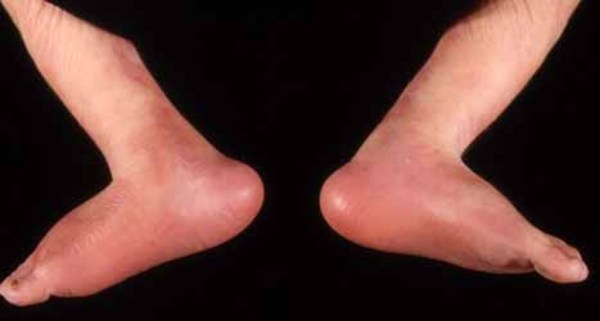
Other skeletal abnormalities that often occur in this disorder include a short, broad neck and chest; prominent shoulder blades; and shoulders that slope less than usual (square shoulders). Affected individuals may have abnormal spinal curvature such as a rounded upper back that also curves to the side (kyphoscoliosis) or exaggerated curvature of the lower back (hyperlordosis). People with 3-M syndrome can also have unusual curving of the fingers (clinodactyly), short fifth (pinky) fingers, prominent heels, and loose joints. Additional skeletal abnormalities, such as unusually slender long bones in the arms and legs; tall, narrow spinal bones (vertebrae); or slightly delayed bone age may be apparent in x-ray images.
A variant of 3-M syndrome called Yakut short stature syndrome has been identified in the isolated Yakut population in the Russian province of Siberia. In addition to having most of the physical features characteristic of 3-M syndrome, people with this form of the disorder are often born with breathing problems that can be life-threatening in infancy.
Frequency
The prevalence of 3-M syndrome is unknown. About 100 individuals worldwide with this disorder have been described in the medical literature.
Causes
Mutations in the CUL7 gene cause 3-M syndrome in more than three-quarters of affected individuals, including those in the Yakut population. Mutations in the OBSL1 gene cause about 16 percent of cases of this disorder. Mutations in other genes, some of which have not been identified, account for the remaining cases.
The CUL7 gene provides instructions for making a protein called cullin-7. The cullin-7 protein plays a role in the cell machinery that breaks down (degrades) unwanted proteins, called the ubiquitin-proteasome system. Specifically, cullin-7 helps assemble a complex called an E3 ubiquitin ligase, which tags the unneeded proteins for degradation. The protein produced from the OBSL1 gene is thought to help maintain normal levels of cullin-7.
The ubiquitin-proteasome system helps regulate the level of proteins involved in several critical cell activities such as the timing of cell division and growth. In particular, the proteins produced from the genes associated with 3-M syndrome are thought to help regulate proteins involved in the body's response to growth hormones, although their specific role in this process is unknown.
Mutations in the CUL7 or OBSL1 gene prevent the cullin-7 protein from bringing together the components of the E3 ubiquitin ligase complex, interfering with the process of tagging unneeded proteins for degradation. The body's response to growth hormones may be impaired as a result. However, the specific relationship between CUL7 and OBSL1 gene mutations and the signs and symptoms of 3-M syndrome are unknown.
Inheritance
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.

Other Names for This Condition
- 3-MSBN
- Dolichospondylic dysplasia
- Le Merrer syndrome
- Three M syndrome
- Three-M slender-boned nanism
- Yakut short stature syndrome
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Clayton PE, Hanson D, Magee L, Murray PG, Saunders E, Abu-Amero SN, Moore GE, Black GC. Exploring the spectrum of 3-M syndrome, a primordial short stature disorder of disrupted ubiquitination. Clin Endocrinol (Oxf). 2012 Sep;77(3):335-42. doi: 10.1111/j.1365-2265.2012.04428.x. Citation on PubMed
- Hanson D, Murray PG, Black GC, Clayton PE. The genetics of 3-M syndrome: unravelling a potential new regulatory growth pathway. Horm Res Paediatr. 2011;76(6):369-78. doi: 10.1159/000334392. Epub 2011 Nov 29. Citation on PubMed
- Hanson D, Murray PG, Coulson T, Sud A, Omokanye A, Stratta E, Sakhinia F, Bonshek C, Wilson LC, Wakeling E, Temtamy SA, Aglan M, Rosser EM, Mansour S, Carcavilla A, Nampoothiri S, Khan WI, Banerjee I, Chandler KE, Black GC, Clayton PE. Mutations in CUL7, OBSL1 and CCDC8 in 3-M syndrome lead to disordered growth factor signalling. J Mol Endocrinol. 2012 Oct 30;49(3):267-75. doi: 10.1530/JME-12-0034. Print 2012 Dec. Citation on PubMed
- Hanson D, Murray PG, O'Sullivan J, Urquhart J, Daly S, Bhaskar SS, Biesecker LG, Skae M, Smith C, Cole T, Kirk J, Chandler K, Kingston H, Donnai D, Clayton PE, Black GC. Exome sequencing identifies CCDC8 mutations in 3-M syndrome, suggesting that CCDC8 contributes in a pathway with CUL7 and OBSL1 to control human growth. Am J Hum Genet. 2011 Jul 15;89(1):148-53. doi: 10.1016/j.ajhg.2011.05.028. Epub 2011 Jul 7. Citation on PubMed or Free article on PubMed Central
- Hanson D, Murray PG, Sud A, Temtamy SA, Aglan M, Superti-Furga A, Holder SE, Urquhart J, Hilton E, Manson FD, Scambler P, Black GC, Clayton PE. The primordial growth disorder 3-M syndrome connects ubiquitination to the cytoskeletal adaptor OBSL1. Am J Hum Genet. 2009 Jun;84(6):801-6. doi: 10.1016/j.ajhg.2009.04.021. Epub 2009 May 28. Citation on PubMed or Free article on PubMed Central
- Huber C, Dias-Santagata D, Glaser A, O'Sullivan J, Brauner R, Wu K, Xu X, Pearce K, Wang R, Uzielli ML, Dagoneau N, Chemaitilly W, Superti-Furga A, Dos Santos H, Megarbane A, Morin G, Gillessen-Kaesbach G, Hennekam R, Van der Burgt I, Black GC, Clayton PE, Read A, Le Merrer M, Scambler PJ, Munnich A, Pan ZQ, Winter R, Cormier-Daire V. Identification of mutations in CUL7 in 3-M syndrome. Nat Genet. 2005 Oct;37(10):1119-24. doi: 10.1038/ng1628. Epub 2005 Sep 4. Citation on PubMed
- Huber C, Fradin M, Edouard T, Le Merrer M, Alanay Y, Da Silva DB, David A, Hamamy H, van Hest L, Lund AM, Michaud J, Oley C, Patel C, Rajab A, Skidmore DL, Stewart H, Tauber M, Munnich A, Cormier-Daire V. OBSL1 mutations in 3-M syndrome are associated with a modulation of IGFBP2 and IGFBP5 expression levels. Hum Mutat. 2010 Jan;31(1):20-6. doi: 10.1002/humu.21150. Citation on PubMed
- Maksimova N, Hara K, Miyashia A, Nikolaeva I, Shiga A, Nogovicina A, Sukhomyasova A, Argunov V, Shvedova A, Ikeuchi T, Nishizawa M, Kuwano R, Onodera O. Clinical, molecular and histopathological features of short stature syndrome with novel CUL7 mutation in Yakuts: new population isolate in Asia. J Med Genet. 2007 Dec;44(12):772-8. doi: 10.1136/jmg.2007.051979. Epub 2007 Aug 3. Citation on PubMed or Free article on PubMed Central
- Temtamy SA, Aglan MS, Ashour AM, Ramzy MI, Hosny LA, Mostafa MI. 3-M syndrome: a report of three Egyptian cases with review of the literature. Clin Dysmorphol. 2006 Apr;15(2):55-64. doi: 10.1097/01.mcd.0000198926.01706.33. Citation on PubMed
- van der Wal G, Otten BJ, Brunner HG, van der Burgt I. 3-M syndrome: description of six new patients with review of the literature. Clin Dysmorphol. 2001 Oct;10(4):241-52. doi: 10.1097/00019605-200110000-00002. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.