

Description
22q13.3 deletion syndrome, which is also known as Phelan-McDermid syndrome, is a disorder caused by the loss of a small piece of chromosome 22. The deletion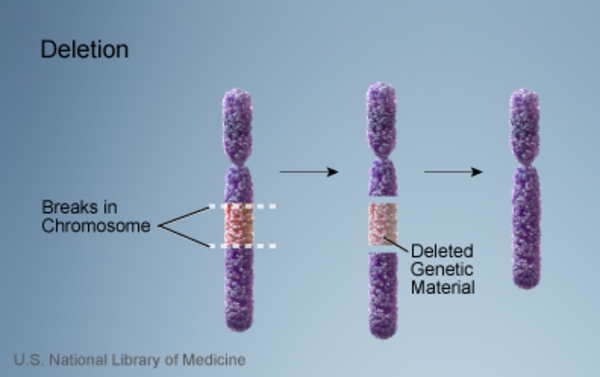 occurs near the end of the chromosome at a location designated q13.3.
occurs near the end of the chromosome at a location designated q13.3.
The features of 22q13.3 deletion syndrome vary widely and involve many parts of the body. Characteristic signs and symptoms include developmental delay, moderate to profound intellectual disability, decreased muscle tone (hypotonia), and absent or delayed speech. Some people with this condition have autism spectrum disorder or autistic-like characteristics that affects communication and social interaction, such as poor eye contact, sensitivity to touch, and aggression. They may also chew on non-food items such as clothing. Less frequently, people with this condition have seizures or lose skills they had already acquired (developmental regression).
Individuals with 22q13.3 deletion syndrome tend to have a decreased sensitivity to pain. Many also have a reduced ability to sweat, which can lead to a greater risk of overheating and dehydration. Some people with this condition have episodes of frequent vomiting and nausea (cyclic vomiting) and backflow of stomach acids into the esophagus (gastroesophageal reflux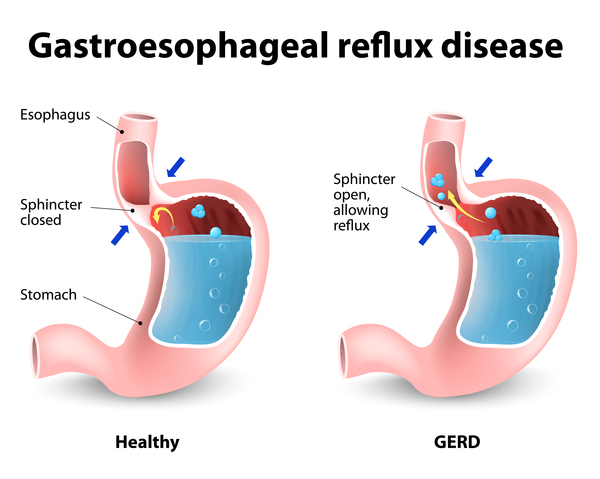 ).
).
People with 22q13.3 deletion syndrome typically have distinctive facial features, including a long, narrow head; prominent ears; a pointed chin ; droopy eyelids (ptosis
; droopy eyelids (ptosis ); and deep-set eyes
); and deep-set eyes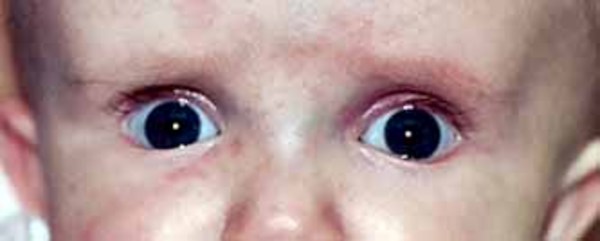 . Other physical features seen with this condition include large and fleshy hands and/or feet, a fusion of the second and third toes (syndactyly
. Other physical features seen with this condition include large and fleshy hands and/or feet, a fusion of the second and third toes (syndactyly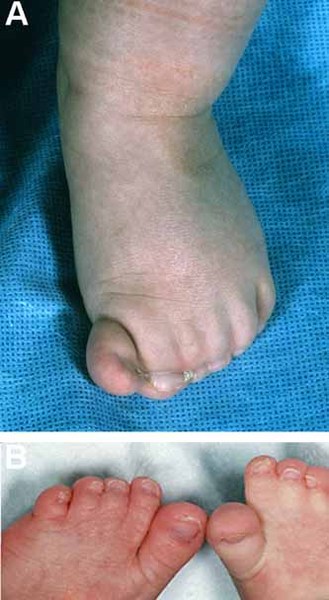 ), and small or abnormal toenails. Some affected individuals have rapid (accelerated) growth.
), and small or abnormal toenails. Some affected individuals have rapid (accelerated) growth.
Frequency
More than 2,200 people have been diagnosed with 22q13.3 deletion syndrome worldwide.
Causes
22q13.3 deletion syndrome is caused by a deletion near the end of the long (q) arm of chromosome 22. The signs and symptoms of 22q13.3 deletion syndrome are probably related to the loss of multiple genes in this region. The size of the deletion varies among affected individuals.
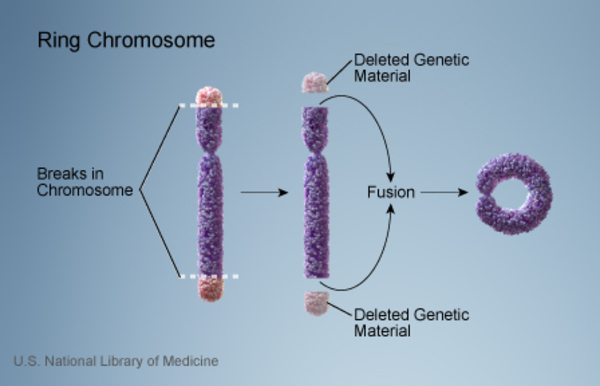
A ring chromosome 22 can also cause 22q13.3 deletion syndrome. A ring chromosome is a circular structure that occurs when a chromosome breaks in two places, the tips of the chromosome are lost, and the broken ends fuse together. People with ring chromosome 22 have one copy of this abnormal chromosome in some or all of their cells. Researchers believe that several critical genes near the end of the long (q) arm of chromosome 22 are lost when the ring chromosome 22 forms. If one of the chromosome break points is at position 22q13.3, people with ring chromosome 22 have similar signs and symptoms as those with a simple deletion.
Researchers are working to identify all of the genes that contribute to the features of 22q13.3 deletion syndrome. They have determined that the loss of a particular gene on chromosome 22, SHANK3, is likely to be responsible for many of the syndrome's characteristic signs (such as developmental delay, intellectual disability, and impaired speech). Additional genes in the deleted region probably contribute to the varied features of 22q13.3 deletion syndrome.
Inheritance
Most cases of 22q13.3 deletion syndrome are not inherited. The deletion occurs most often as a random event during the formation of reproductive cells (eggs or sperm) or in early fetal development. Affected people typically have no history of the disorder in their family, though they can pass the chromosome deletion to their children.
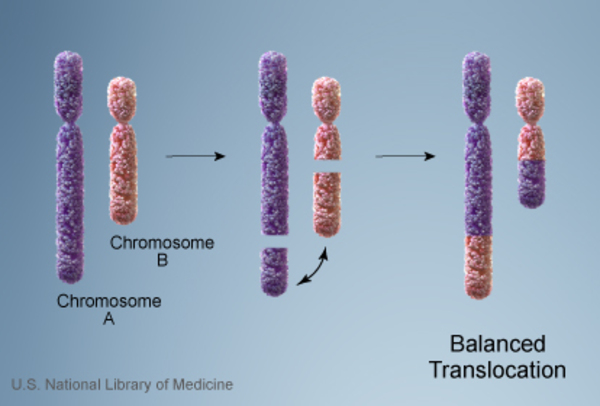
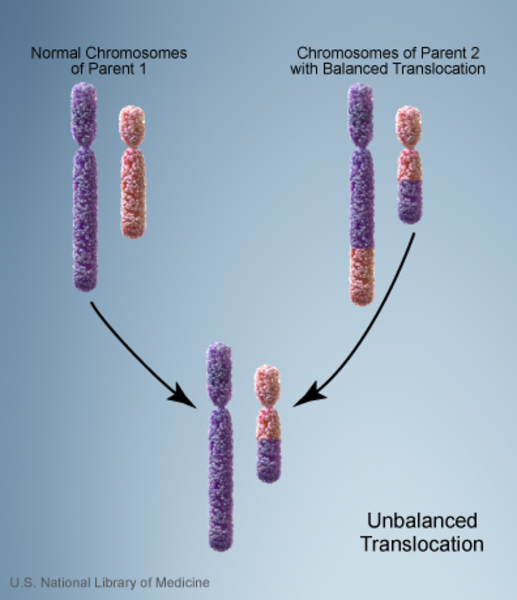
When 22q13.3 deletion syndrome is inherited, its inheritance pattern is considered autosomal dominant because a deletion in one copy of chromosome 22 in each cell is sufficient to cause the condition. About 15 to 20 percent of people with 22q13.3 deletion syndrome inherit a chromosome abnormality from an unaffected parent. In these cases, the parent carries a chromosomal rearrangement called a balanced translocation, in which a segment from one chromosome has traded places with a segment from another chromosome, but no genetic material is gained or lost. Balanced translocations usually do not cause any health problems; however, they can become unbalanced as they are passed to the next generation. Children who inherit an unbalanced translocation can have a chromosomal rearrangement with extra or missing genetic material. Individuals with 22q13.3 deletion syndrome who inherit an unbalanced translocation are missing genetic material from the long arm of chromosome 22, which results in the health problems characteristic of this disorder.

Other Names for This Condition
- 22q13 deletion syndrome
- Deletion 22q13 syndrome
- Deletion 22q13.3 syndrome
- Monosomy 22q13
- Phelan-McDermid syndrome
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Bonaglia MC, Giorda R, Mani E, Aceti G, Anderlid BM, Baroncini A, Pramparo T, Zuffardi O. Identification of a recurrent breakpoint within the SHANK3 gene in the 22q13.3 deletion syndrome. J Med Genet. 2006 Oct;43(10):822-8. doi: 10.1136/jmg.2005.038604. Epub 2005 Nov 11. Citation on PubMed or Free article on PubMed Central
- Cusmano-Ozog K, Manning MA, Hoyme HE. 22q13.3 deletion syndrome: a recognizable malformation syndrome associated with marked speech and language delay. Am J Med Genet C Semin Med Genet. 2007 Nov 15;145C(4):393-8. doi: 10.1002/ajmg.c.30155. Citation on PubMed
- Havens JM, Visootsak J, Phelan MC, Graham JM Jr. 22q13 deletion syndrome: an update and review for the primary pediatrician. Clin Pediatr (Phila). 2004 Jan-Feb;43(1):43-53. doi: 10.1177/000992280404300106. Citation on PubMed
- Koolen DA, Reardon W, Rosser EM, Lacombe D, Hurst JA, Law CJ, Bongers EM, van Ravenswaaij-Arts CM, Leisink MA, van Kessel AG, Veltman JA, de Vries BB. Molecular characterisation of patients with subtelomeric 22q abnormalities using chromosome specific array-based comparative genomic hybridisation. Eur J Hum Genet. 2005 Sep;13(9):1019-24. doi: 10.1038/sj.ejhg.5201456. Citation on PubMed
- Manning MA, Cassidy SB, Clericuzio C, Cherry AM, Schwartz S, Hudgins L, Enns GM, Hoyme HE. Terminal 22q deletion syndrome: a newly recognized cause of speech and language disability in the autism spectrum. Pediatrics. 2004 Aug;114(2):451-7. doi: 10.1542/peds.114.2.451. Citation on PubMed
- Phelan MC, Rogers RC, Saul RA, Stapleton GA, Sweet K, McDermid H, Shaw SR, Claytor J, Willis J, Kelly DP. 22q13 deletion syndrome. Am J Med Genet. 2001 Jun 15;101(2):91-9. doi: 10.1002/1096-8628(20010615)101:23.0.co;2-c. Citation on PubMed
- Phelan MC. Deletion 22q13.3 syndrome. Orphanet J Rare Dis. 2008 May 27;3:14. doi: 10.1186/1750-1172-3-14. Citation on PubMed or Free article on PubMed Central
- Wilson HL, Wong AC, Shaw SR, Tse WY, Stapleton GA, Phelan MC, Hu S, Marshall J, McDermid HE. Molecular characterisation of the 22q13 deletion syndrome supports the role of haploinsufficiency of SHANK3/PROSAP2 in the major neurological symptoms. J Med Genet. 2003 Aug;40(8):575-84. doi: 10.1136/jmg.40.8.575. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.