

Description
16p11.2 deletion syndrome is a disorder caused by a deletion of a small piece of chromosome 16. The deletion occurs near the middle of the chromosome at a location designated p11.2.
People with 16p11.2 deletion syndrome usually have developmental delay and intellectual disability. Most also have at least some features of autism spectrum disorders. These disorders are characterized by impaired communication and socialization skills, as well as delayed development of speech and language. In 16p11.2 deletion syndrome, expressive language skills (vocabulary and the production of speech) are generally more severely affected than receptive language skills (the ability to understand speech). Some people with this disorder have recurrent seizures (epilepsy).
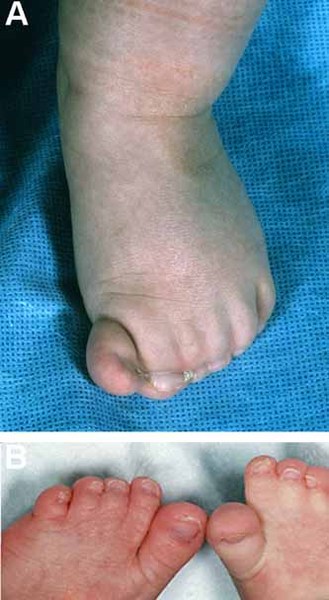
Some affected individuals have minor physical abnormalities such as low-set ears or partially webbed toes (partial syndactyly). People with this disorder are also at increased risk of obesity compared with the general population. However, there is no particular pattern of physical abnormalities that characterizes 16p11.2 deletion syndrome. Signs and symptoms of the disorder vary even among affected members of the same family. Some people with the deletion have no identified physical, intellectual, or behavioral abnormalities.
Frequency
Most people tested for the 16p11.2 deletion have come to medical attention as a result of developmental delay or autistic characteristics. Other individuals with the 16p11.2 deletion have no associated health or behavioral problems, and so the deletion may never be detected. For this reason, the prevalence of this deletion in the general population is difficult to determine but has been estimated at approximately 3 in 10,000.
Causes
People with 16p11.2 deletion syndrome are missing a sequence of about 600,000 DNA building blocks (base pairs), also written as 600 kilobases (kb), at position p11.2 on chromosome 16. This deletion affects one of the two copies of chromosome 16 in each cell. The 600 kb region contains more than 25 genes, and in many cases little is known about their function. Researchers are working to determine how the missing genes contribute to the features of 16p11.2 deletion syndrome.
Inheritance
16p11.2 deletion syndrome is considered to have an autosomal dominant inheritance pattern because a deletion in one copy of chromosome 16 in each cell is sufficient to cause the condition. However, most cases of 16p11.2 deletion syndrome are not inherited. The deletion occurs most often as a random event during the formation of reproductive cells (eggs and sperm) or in early fetal development. Affected people typically have no history of the disorder in their family, although they can pass the condition to their children. Several examples of inherited 16p11.2 deletion have been reported. In inherited cases, other family members may be affected as well.


Other Names for This Condition
- Autism, susceptibility to, 14A
- AUTS14A
Additional Information & Resources
Genetic Testing Information
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Bijlsma EK, Gijsbers AC, Schuurs-Hoeijmakers JH, van Haeringen A, Fransen van de Putte DE, Anderlid BM, Lundin J, Lapunzina P, Perez Jurado LA, Delle Chiaie B, Loeys B, Menten B, Oostra A, Verhelst H, Amor DJ, Bruno DL, van Essen AJ, Hordijk R, Sikkema-Raddatz B, Verbruggen KT, Jongmans MC, Pfundt R, Reeser HM, Breuning MH, Ruivenkamp CA. Extending the phenotype of recurrent rearrangements of 16p11.2: deletions in mentally retarded patients without autism and in normal individuals. Eur J Med Genet. 2009 Mar-Jun;52(2-3):77-87. doi: 10.1016/j.ejmg.2009.03.006. Epub 2009 Mar 21. Citation on PubMed
- Ciuladaite Z, Kasnauskiene J, Cimbalistiene L, Preiksaitiene E, Patsalis PC, Kucinskas V. Mental retardation and autism associated with recurrent 16p11.2 microdeletion: incomplete penetrance and variable expressivity. J Appl Genet. 2011 Nov;52(4):443-9. doi: 10.1007/s13353-011-0063-z. Epub 2011 Sep 20. No abstract available. Citation on PubMed
- Fernandez BA, Roberts W, Chung B, Weksberg R, Meyn S, Szatmari P, Joseph-George AM, Mackay S, Whitten K, Noble B, Vardy C, Crosbie V, Luscombe S, Tucker E, Turner L, Marshall CR, Scherer SW. Phenotypic spectrum associated with de novo and inherited deletions and duplications at 16p11.2 in individuals ascertained for diagnosis of autism spectrum disorder. J Med Genet. 2010 Mar;47(3):195-203. doi: 10.1136/jmg.2009.069369. Epub 2009 Sep 15. Citation on PubMed
- Ghebranious N, Giampietro PF, Wesbrook FP, Rezkalla SH. A novel microdeletion at 16p11.2 harbors candidate genes for aortic valve development, seizure disorder, and mild mental retardation. Am J Med Genet A. 2007 Jul 1;143A(13):1462-71. doi: 10.1002/ajmg.a.31837. Citation on PubMed
- Hanson E, Bernier R, Porche K, Jackson FI, Goin-Kochel RP, Snyder LG, Snow AV, Wallace AS, Campe KL, Zhang Y, Chen Q, D'Angelo D, Moreno-De-Luca A, Orr PT, Boomer KB, Evans DW, Kanne S, Berry L, Miller FK, Olson J, Sherr E, Martin CL, Ledbetter DH, Spiro JE, Chung WK; Simons Variation in Individuals Project Consortium. The cognitive and behavioral phenotype of the 16p11.2 deletion in a clinically ascertained population. Biol Psychiatry. 2015 May 1;77(9):785-93. doi: 10.1016/j.biopsych.2014.04.021. Epub 2014 Jun 16. Citation on PubMed
- Kumar RA, KaraMohamed S, Sudi J, Conrad DF, Brune C, Badner JA, Gilliam TC, Nowak NJ, Cook EH Jr, Dobyns WB, Christian SL. Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet. 2008 Feb 15;17(4):628-38. doi: 10.1093/hmg/ddm376. Epub 2007 Dec 21. Citation on PubMed
- Shimojima K, Inoue T, Fujii Y, Ohno K, Yamamoto T. A familial 593-kb microdeletion of 16p11.2 associated with mental retardation and hemivertebrae. Eur J Med Genet. 2009 Nov-Dec;52(6):433-5. doi: 10.1016/j.ejmg.2009.09.007. Epub 2009 Sep 19. Citation on PubMed
- Taylor CM, Smith R, Lehman C, Mitchel MW, Singer K, Weaver WC, Chung W. 16p11.2 Recurrent Deletion. 2009 Sep 22 [updated 2021 Oct 28]. In: Adam MP, Bick S, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2026. Available from http://www.ncbi.nlm.nih.gov/books/NBK11167/ Citation on PubMed
- Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, Saemundsen E, Stefansson H, Ferreira MA, Green T, Platt OS, Ruderfer DM, Walsh CA, Altshuler D, Chakravarti A, Tanzi RE, Stefansson K, Santangelo SL, Gusella JF, Sklar P, Wu BL, Daly MJ; Autism Consortium. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008 Feb 14;358(7):667-75. doi: 10.1056/NEJMoa075974. Epub 2008 Jan 9. Citation on PubMed
- Zufferey F, Sherr EH, Beckmann ND, Hanson E, Maillard AM, Hippolyte L, Mace A, Ferrari C, Kutalik Z, Andrieux J, Aylward E, Barker M, Bernier R, Bouquillon S, Conus P, Delobel B, Faucett WA, Goin-Kochel RP, Grant E, Harewood L, Hunter JV, Lebon S, Ledbetter DH, Martin CL, Mannik K, Martinet D, Mukherjee P, Ramocki MB, Spence SJ, Steinman KJ, Tjernagel J, Spiro JE, Reymond A, Beckmann JS, Chung WK, Jacquemont S; Simons VIP Consortium; 16p11.2 European Consortium. A 600 kb deletion syndrome at 16p11.2 leads to energy imbalance and neuropsychiatric disorders. J Med Genet. 2012 Oct;49(10):660-8. doi: 10.1136/jmedgenet-2012-101203. Citation on PubMed or Free article on PubMed Central
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.