

Description
Swyer syndrome is a condition that affects sex development. Sex development usually follows a particular path based on an individual's chromosomes; however, in Swyer syndrome, sex development is not typical for the affected individual's chromosomal pattern.
Chromosomes contain the genetic instructions for how the body develops and functions. People usually have 46 chromosomes in each cell. Two of the 46 chromosomes, known as X and Y, are called sex chromosomes because they help determine whether a person will develop male or female reproductive structures. Girls and women typically have two X chromosomes (46,XX karyotype), while boys and men typically have one X chromosome and one Y chromosome (46,XY karyotype
because they help determine whether a person will develop male or female reproductive structures. Girls and women typically have two X chromosomes (46,XX karyotype), while boys and men typically have one X chromosome and one Y chromosome (46,XY karyotype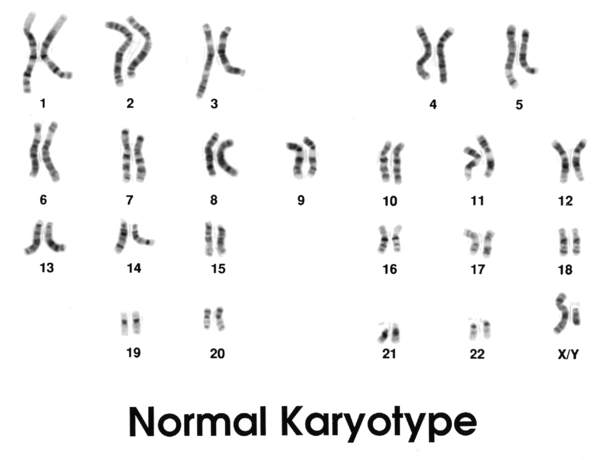 ). In Swyer syndrome, individuals have one X chromosome and one Y chromosome in each cell, which is the pattern typically found in boys and men; however, they have female reproductive structures
). In Swyer syndrome, individuals have one X chromosome and one Y chromosome in each cell, which is the pattern typically found in boys and men; however, they have female reproductive structures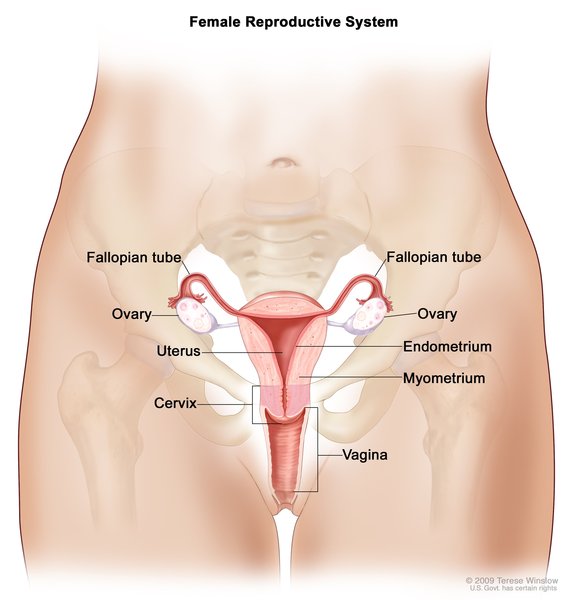 .
.
People with Swyer syndrome have female external genitalia and some female internal reproductive structures. These individuals usually have a uterus and fallopian tubes, but their gonads (ovaries or testes) are not functional. Instead, the gonads are small and underdeveloped and contain little gonadal tissue. These structures are called streak gonads. The streak gonadal tissue is at risk of developing cancer that is often hard-to-detect, so it is usually removed surgically. Swyer syndrome is also called 46,XY complete gonadal dysgenesis; the medical term “dysgenesis” means "abnormal development."
Because they appear female on the outside, babies with Swyer syndrome are usually raised as girls and develop a female gender identity, which is a person's sense of their gender (girl, boy, a combination, or neither). Swyer syndrome may be identified before birth, at birth, or later when a child does not go through puberty as usual. Because they do not have functional ovaries that produce hormones, affected individuals often begin hormone replacement therapy during early adolescence to start puberty, causing the breasts and uterus to grow, and eventually leading to menstruation. Hormone replacement therapy is also important for bone health and helps reduce the risk of low bone density (osteopenia) and fragile bones (osteoporosis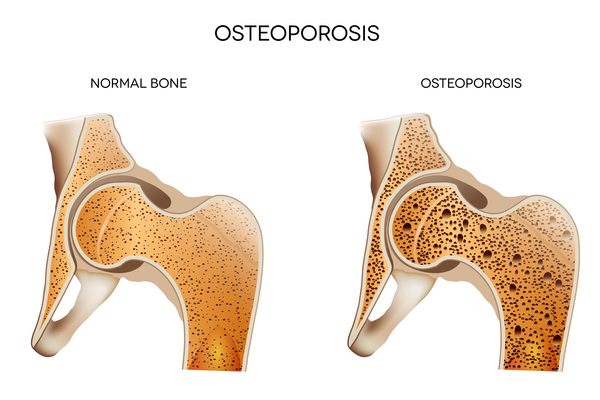 ). Women with Swyer syndrome do not produce eggs (ova), but if they have a uterus, they may be able to become pregnant with a donated egg or embryo.
). Women with Swyer syndrome do not produce eggs (ova), but if they have a uterus, they may be able to become pregnant with a donated egg or embryo.
Frequency
Swyer syndrome occurs in approximately 1 in 80,000 people.
Causes
In many individuals with Swyer syndrome, the cause is unknown. However, variants (also known as mutations) in one of several genes have been found to cause the condition in some affected individuals.
Variants in the SRY gene have been found in approximately 15 percent of individuals with Swyer syndrome. The SRY gene, located on the Y chromosome, provides instructions for making a protein called sex-determining region Y. This protein attaches (binds) to specific regions of DNA and helps control the activity of particular genes. The sex-determining region Y protein starts processes that are involved in male-typical sex development. These processes cause a fetus to develop male gonads (testes) and genitals and prevent the development of female internal reproductive structures (uterus, fallopian tubes, and upper part of the vagina) and genitals. SRY gene variants that cause Swyer syndrome prevent production of the sex-determining region Y protein or result in the production of a nonfunctioning protein. Without functional sex-determining region Y protein, a fetus will not develop testes but will develop female-typical internal and external reproductive structures, despite having an X and a Y chromosome.
Swyer syndrome can also be caused by variants in the MAP3K1 gene; research indicates that variants in this gene may account for up to 18 percent of cases. The MAP3K1 gene provides instructions for making a protein that helps control various processes in the body, including processes of determining sex characteristics before birth. The variants in the MAP3K1 gene that cause Swyer syndrome decrease signaling that leads to male-typical sex development and increase signaling that leads to female-typical sex development. These changes in signaling prevent the development of testes and allow the development of female reproductive structures.
Variants in the DHH and NR5A1 genes have also been identified in a small percentage of people with Swyer syndrome. The DHH gene provides instructions for making a protein that is important for early development of tissues in many parts of the body. The NR5A1 gene provides instructions for producing a protein called steroidogenic factor 1 (SF1). This protein helps control the activity of several genes related to sex development and the production of sex hormones. Variants in the DHH and NR5A1 genes disrupt the process of sex development, preventing affected individuals with a 46,XY karyotype from developing testes and causing them to develop female reproductive structures.
Changes affecting other genes have also been identified in a few people with Swyer syndrome. Nongenetic factors, such as hormonal medications taken by the mother during pregnancy, have very rarely been associated with this condition.
Inheritance
Most cases of Swyer syndrome are not inherited; they occur in people with no history of the condition in their family. These cases often result from new (de novo) variants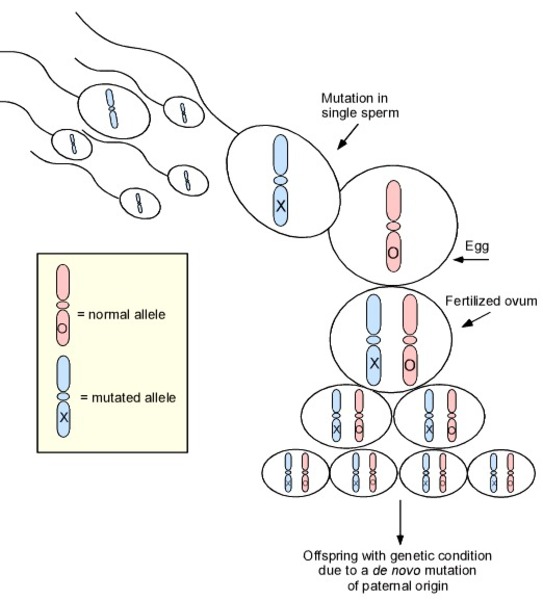 in a gene that occur during the formation of reproductive cells (eggs or sperm) or in early embryonic development. Noninherited cases can rarely result from nongenetic causes.
in a gene that occur during the formation of reproductive cells (eggs or sperm) or in early embryonic development. Noninherited cases can rarely result from nongenetic causes.
SRY-related Swyer syndrome is usually caused by a new variant that is not inherited from either parent. However, some individuals with Swyer syndrome inherit an altered SRY gene from an unaffected father who is mosaic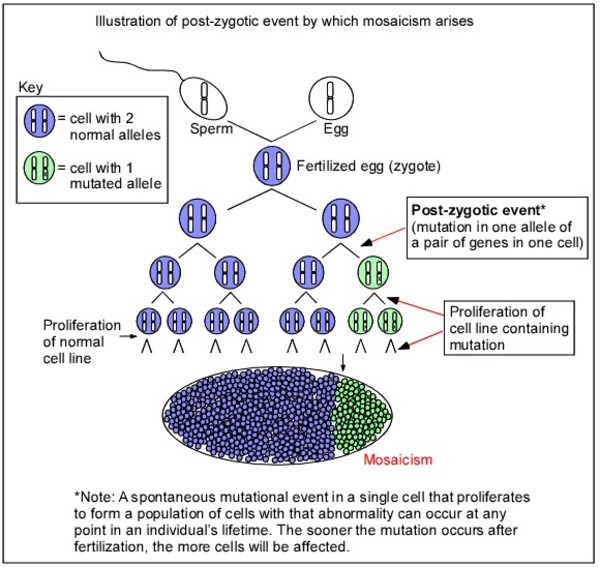 for the variant. Mosaic means that an individual has the variant in some cells (which can include some reproductive cells) but not in others. Because he has some cells that do not have the genetic variant, he does not have the condition and is able to have children; however, he can pass the variant to his offspring. In rare cases, a father may carry the variant in every cell of the body but also has other genetic variations that prevent him from being affected by the condition. Because the SRY gene is on the Y chromosome, Swyer syndrome caused by SRY gene variants is described as having a Y-linked inheritance pattern
for the variant. Mosaic means that an individual has the variant in some cells (which can include some reproductive cells) but not in others. Because he has some cells that do not have the genetic variant, he does not have the condition and is able to have children; however, he can pass the variant to his offspring. In rare cases, a father may carry the variant in every cell of the body but also has other genetic variations that prevent him from being affected by the condition. Because the SRY gene is on the Y chromosome, Swyer syndrome caused by SRY gene variants is described as having a Y-linked inheritance pattern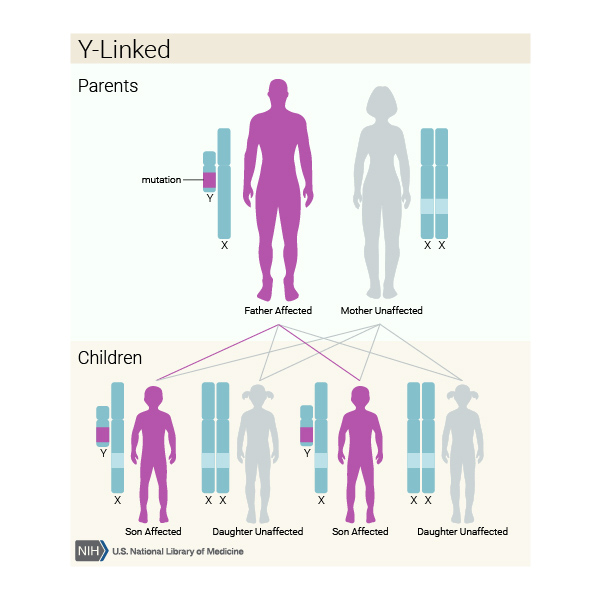 .
.
When Swyer syndrome is associated with an MAP3K1 or NR5A1 gene variant, the condition is also often caused by a new variant that is not inherited. In the rare inherited cases, the variant may be inherited from either parent, because these genes are not on the Y chromosome; however, only children with an XY chromosome pattern are affected (the condition is said to be sex-limited). In inherited cases, the condition has an autosomal dominant pattern of inheritance, which means one copy of the altered gene in each cell is sufficient to cause the condition. However, the parent with the genetic variant typically does not have signs and symptoms.
of inheritance, which means one copy of the altered gene in each cell is sufficient to cause the condition. However, the parent with the genetic variant typically does not have signs and symptoms.
Swyer syndrome caused by variants in the DHH gene is inherited in a sex-limited autosomal recessive pattern , which means both copies of the gene in each cell have variants, and only individuals with an XY chromosome pattern are affected. The parents of an individual with an autosomal recessive condition each have one copy
, which means both copies of the gene in each cell have variants, and only individuals with an XY chromosome pattern are affected. The parents of an individual with an autosomal recessive condition each have one copy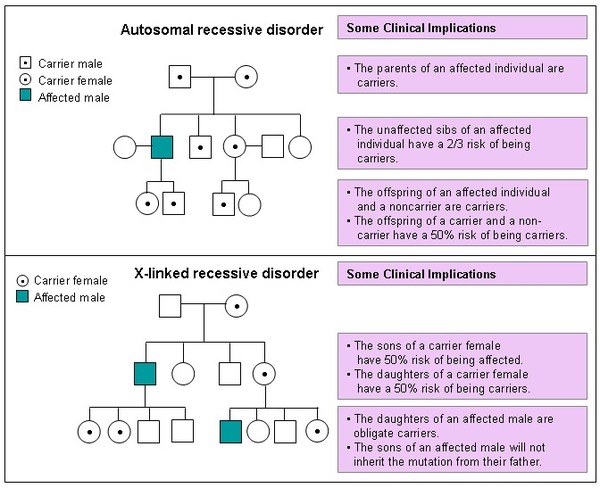 of the altered gene, and they typically do not have signs and symptoms of the condition.
of the altered gene, and they typically do not have signs and symptoms of the condition.
Other Names for This Condition
- 46,XY CGD
- 46,XY complete gonadal dysgenesis
- 46,XY sex reversal
- Gonadal dysgenesis, 46,XY
- Pure gonadal dysgenesis 46,XY
- XY pure gonadal dysgenesis
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Scientific Articles on PubMed
References
- Arboleda VA, Sandberg DE, Vilain E. DSDs: genetics, underlying pathologies and psychosexual differentiation. Nat Rev Endocrinol. 2014 Oct;10(10):603-15. doi: 10.1038/nrendo.2014.130. Epub 2014 Aug 5. Citation on PubMed or Free article on PubMed Central
- Baxter RM, Arboleda VA, Lee H, Barseghyan H, Adam MP, Fechner PY, Bargman R, Keegan C, Travers S, Schelley S, Hudgins L, Mathew RP, Stalker HJ, Zori R, Gordon OK, Ramos-Platt L, Pawlikowska-Haddal A, Eskin A, Nelson SF, Delot E, Vilain E. Exome sequencing for the diagnosis of 46,XY disorders of sex development. J Clin Endocrinol Metab. 2015 Feb;100(2):E333-44. doi: 10.1210/jc.2014-2605. Epub 2014 Nov 10. Citation on PubMed
- Baxter RM, Vilain E. Translational genetics for diagnosis of human disorders of sex development. Annu Rev Genomics Hum Genet. 2013;14:371-92. doi: 10.1146/annurev-genom-091212-153417. Epub 2013 Jul 15. Citation on PubMed or Free article on PubMed Central
- Delot EC, Vilain E. Towards improved genetic diagnosis of human differences of sex development. Nat Rev Genet. 2021 Sep;22(9):588-602. doi: 10.1038/s41576-021-00365-5. Epub 2021 Jun 3. Citation on PubMed
- El-Khairi R, Achermann JC. Steroidogenic factor-1 and human disease. Semin Reprod Med. 2012 Oct;30(5):374-81. doi: 10.1055/s-0032-1324720. Epub 2012 Oct 8. Citation on PubMed
- Fabbri-Scallet H, de Sousa LM, Maciel-Guerra AT, Guerra-Junior G, de Mello MP. Mutation update for the NR5A1 gene involved in DSD and infertility. Hum Mutat. 2020 Jan;41(1):58-68. doi: 10.1002/humu.23916. Epub 2019 Sep 27. Citation on PubMed
- Granados A, Alaniz VI, Mohnach L, Barseghyan H, Vilain E, Ostrer H, Quint EH, Chen M, Keegan CE. MAP3K1-related gonadal dysgenesis: Six new cases and review of the literature. Am J Med Genet C Semin Med Genet. 2017 Jun;175(2):253-259. doi: 10.1002/ajmg.c.31559. Epub 2017 May 15. Citation on PubMed
- King TF, Conway GS. Swyer syndrome. Curr Opin Endocrinol Diabetes Obes. 2014 Dec;21(6):504-10. doi: 10.1097/MED.0000000000000113. Citation on PubMed
- Loke J, Pearlman A, Radi O, Zuffardi O, Giussani U, Pallotta R, Camerino G, Ostrer H. Mutations in MAP3K1 tilt the balance from SOX9/FGF9 to WNT/beta-catenin signaling. Hum Mol Genet. 2014 Feb 15;23(4):1073-83. doi: 10.1093/hmg/ddt502. Epub 2013 Oct 16. Citation on PubMed
- Massanyi EZ, Dicarlo HN, Migeon CJ, Gearhart JP. Review and management of 46,XY disorders of sex development. J Pediatr Urol. 2013 Jun;9(3):368-79. doi: 10.1016/j.jpurol.2012.12.002. Epub 2012 Dec 29. Citation on PubMed
- McElreavey K, Jorgensen A, Eozenou C, Merel T, Bignon-Topalovic J, Tan DS, Houzelstein D, Buonocore F, Warr N, Kay RGG, Peycelon M, Siffroi JP, Mazen I, Achermann JC, Shcherbak Y, Leger J, Sallai A, Carel JC, Martinerie L, Le Ru R, Conway GS, Mignot B, Van Maldergem L, Bertalan R, Globa E, Brauner R, Jauch R, Nef S, Greenfield A, Bashamboo A. Pathogenic variants in the DEAH-box RNA helicase DHX37 are a frequent cause of 46,XY gonadal dysgenesis and 46,XY testicular regression syndrome. Genet Med. 2020 Jan;22(1):150-159. doi: 10.1038/s41436-019-0606-y. Epub 2019 Jul 24. Citation on PubMed
- Michala L, Goswami D, Creighton SM, Conway GS. Swyer syndrome: presentation and outcomes. BJOG. 2008 May;115(6):737-41. doi: 10.1111/j.1471-0528.2008.01703.x. Citation on PubMed
- Mohnach L, Fechner PY, Keegan CE. Nonsyndromic Disorders of Testicular Development Overview. 2008 May 21 [updated 2022 Aug 18]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1547/ Citation on PubMed
- Ostrer H. Disorders of sex development (DSDs): an update. J Clin Endocrinol Metab. 2014 May;99(5):1503-9. doi: 10.1210/jc.2013-3690. Epub 2014 Apr 23. Citation on PubMed
- Patnayak R, Suresh V, Jena A, Rajagopal G, Vijayalakshmi B, Reddy AP, Rukumangadha M, Sachan A. Swyer syndrome: a case report with literature review. JNMA J Nepal Med Assoc. 2012 Apr-Jun;52(186):72-4. Citation on PubMed
- Pearlman A, Loke J, Le Caignec C, White S, Chin L, Friedman A, Warr N, Willan J, Brauer D, Farmer C, Brooks E, Oddoux C, Riley B, Shajahan S, Camerino G, Homfray T, Crosby AH, Couper J, David A, Greenfield A, Sinclair A, Ostrer H. Mutations in MAP3K1 cause 46,XY disorders of sex development and implicate a common signal transduction pathway in human testis determination. Am J Hum Genet. 2010 Dec 10;87(6):898-904. doi: 10.1016/j.ajhg.2010.11.003. Citation on PubMed or Free article on PubMed Central
- Tang R, Liu X, Pan L, Chen R. Novel mutation in FTHL17 gene in pedigree with 46,XY pure gonadal dysgenesis. Fertil Steril. 2019 Jun;111(6):1226-1235.e1. doi: 10.1016/j.fertnstert.2019.01.027. Epub 2019 Mar 25. Citation on PubMed
- Zhu J, Liu X, Jin H, Lu X. Swyer syndrome, 46,XY gonadal dysgenesis, a sex reversal disorder with dysgerminoma: a case report and literature review. Clin Exp Obstet Gynecol. 2011;38(4):414-8. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.