

Description
Nonsyndromic hearing loss is a partial or total loss of hearing that is not associated with other signs and symptoms. In contrast, syndromic hearing loss occurs with signs and symptoms affecting other parts of the body.
Nonsyndromic hearing loss can be classified in several different ways. One common way is by the condition's pattern of inheritance: autosomal dominant (DFNA), autosomal recessive (DFNB), X-linked (DFNX), or mitochondrial (which does not have a special designation). Each of these types of hearing loss includes multiple subtypes. DFNA, DFNB, and DFNX subtypes are numbered in the order in which they were first described. For example, DFNA1 was the first type of autosomal dominant nonsyndromic hearing loss to be identified.
The characteristics of nonsyndromic hearing loss vary among the different types. Hearing loss can affect one ear (unilateral) or both ears (bilateral). Degrees of hearing loss range from mild (difficulty understanding soft speech) to profound (inability to hear even very loud noises). The term "deafness" is often used to describe severe-to-profound hearing loss. Hearing loss can be stable, or it may be progressive, becoming more severe as a person gets older. Particular types of nonsyndromic hearing loss show distinctive patterns of hearing loss. For example, the loss may be more pronounced at high, middle, or low tones.
Most forms of nonsyndromic hearing loss are described as sensorineural, which means they are associated with a permanent loss of hearing caused by damage to structures in the inner ear. The inner ear processes sound and sends the information to the brain in the form of electrical nerve impulses. Less commonly, nonsyndromic hearing loss is described as conductive, meaning it results from changes in the middle ear. The middle ear contains three tiny bones that help transfer sound from the eardrum to the inner ear. Some forms of nonsyndromic hearing loss, particularly a type called DFNX2, involve changes in both the inner ear and the middle ear. This combination is called mixed hearing loss.
Depending on the type, nonsyndromic hearing loss can become apparent at any time from infancy to old age. Hearing loss that is present before a child learns to speak is classified as prelingual or congenital. Hearing loss that occurs after the development of speech is classified as postlingual.
Frequency
Between 2 and 3 per 1,000 children in the United States are born with detectable hearing loss in one or both ears. The prevalence of hearing loss increases with age; the condition affects 1 in 8 people in the United States age 12 and older, or about 30 million people. By age 85, more than half of all people experience hearing loss.
Causes
The causes of nonsyndromic hearing loss are complex. Researchers have identified more than 90 genes that, when altered, are associated with nonsyndromic hearing loss. Many of these genes are involved in the development and function of the inner ear . Mutations in these genes contribute to hearing loss by interfering with critical steps in processing sound. Different mutations in the same gene can be associated with different types of hearing loss, and some genes are associated with both syndromic and nonsyndromic forms. In many affected families, the factors contributing to hearing loss have not been identified.
. Mutations in these genes contribute to hearing loss by interfering with critical steps in processing sound. Different mutations in the same gene can be associated with different types of hearing loss, and some genes are associated with both syndromic and nonsyndromic forms. In many affected families, the factors contributing to hearing loss have not been identified.
Most cases of nonsyndromic hearing loss are inherited in an autosomal recessive pattern. About half of all severe-to-profound autosomal recessive nonsyndromic hearing loss results from mutations in the GJB2 gene; these cases are designated DFNB1. The GJB2 gene provides instructions for making a protein called connexin 26, which is a member of the connexin protein family. Mutations in another connexin gene, GJB6, can also cause DFNB1. The GJB6 gene provides instructions for making a protein called connexin 30. Connexin proteins form channels called gap junctions, which allow communication between neighboring cells, including cells in the inner ear. Mutations in the GJB2 or GJB6 gene alter their respective connexin proteins, which changes the structure of gap junctions and may affect the function or survival of cells that are needed for hearing.
The most common cause of moderate autosomal recessive nonsyndromic hearing loss is mutations in the STRC gene. These mutations cause a form of the condition known as DFNB16. Mutations in more than 60 other genes can also cause autosomal recessive nonsyndromic hearing loss. Many of these gene mutations have been found in one or a few families.
Nonsyndromic hearing loss can also be inherited in an autosomal dominant pattern. Mutations in at least 30 genes have been identified in people with autosomal dominant nonsyndromic hearing loss; mutations in some of these genes (including GJB2 and GJB6) can also cause autosomal recessive forms of the condition. Although no single gene is associated with a majority of autosomal dominant nonsyndromic hearing loss cases, mutations in a few genes, such as KCNQ4 and TECTA, are relatively common. Mutations in many of the other genes associated with autosomal dominant nonsyndromic hearing loss have been found in only one or a few families.
X-linked and mitochondrial forms of nonsyndromic hearing loss are rare. About half of all X-linked cases are caused by mutations in the POU3F4 gene. This form of the condition is designated DFNX2. Mutations in at least three other genes have also been identified in people with X-linked nonsyndromic hearing loss.
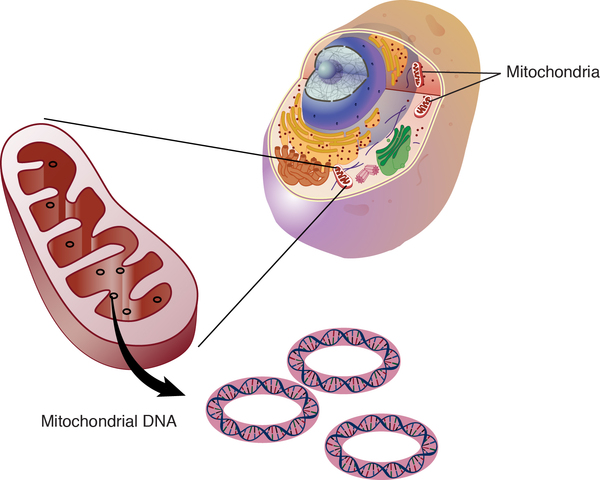
Mitochondrial forms of hearing loss result from changes in mitochondrial DNA (mtDNA). Mitochondria are structures within cells that convert the energy from food into a form that cells can use. Although most DNA is packaged in chromosomes within the nucleus, mitochondria also have a small amount of their own DNA. Only a few mutations in mtDNA have been associated with hearing loss, and their role in the condition is still being studied.
are structures within cells that convert the energy from food into a form that cells can use. Although most DNA is packaged in chromosomes within the nucleus, mitochondria also have a small amount of their own DNA. Only a few mutations in mtDNA have been associated with hearing loss, and their role in the condition is still being studied.
Mutations in some of the genes associated with nonsyndromic hearing loss can also cause syndromic forms of hearing loss, such as Usher syndrome (CDH23 and MYO7A, among others), Pendred syndrome (SLC26A4), Wolfram syndrome (WFS1), and Stickler syndrome (COL11A2). It is often unclear how mutations in the same gene can cause isolated hearing loss in some individuals and hearing loss with additional signs and symptoms in others.
In addition to genetic changes, hearing loss can result from environmental factors or a combination of genetic risk and a person's environmental exposures. Environmental causes of hearing loss include certain medications, specific infections before or after birth, and exposure to loud noise over an extended period. Age is also a major risk factor for hearing loss. Age-related hearing loss (presbycusis) is thought to have both genetic and environmental influences.
Inheritance
As discussed above, nonsyndromic hearing loss has different patterns of inheritance. Between 75 and 80 percent of cases are inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. Usually, each parent of an individual with autosomal recessive hearing loss carries one copy of the mutated gene but does not have hearing loss.

Another 20 to 25 percent of nonsyndromic hearing loss has an autosomal dominant pattern of inheritance, which means one copy of the altered gene in each cell is sufficient to cause the condition. Most people with autosomal dominant hearing loss inherit an altered copy of the gene from a parent who also has hearing loss.

Between 1 and 2 percent of cases have an X-linked pattern of inheritance. A condition is considered X-linked if the mutated gene that causes the disorder is located on the X chromosome, one of the two sex chromosomes in each cell. Males with X-linked nonsyndromic hearing loss tend to develop more severe hearing loss earlier in life than females who inherit a copy of the same gene mutation. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.

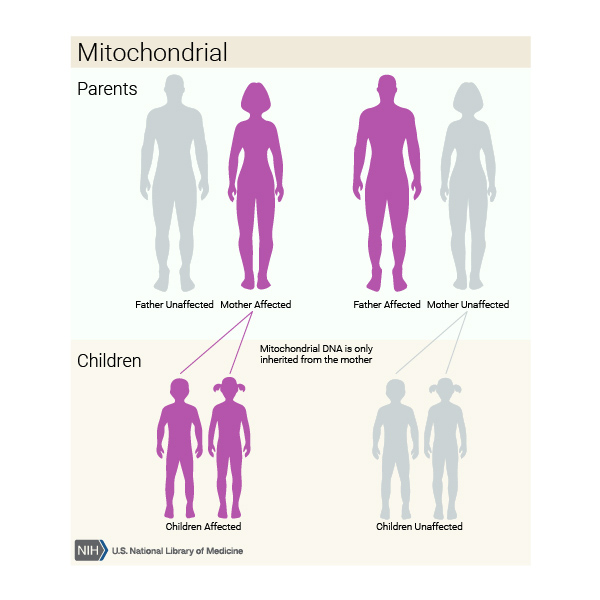
Mitochondrial forms of the condition, which result from changes to mtDNA, account for less than 1 percent of all nonsyndromic hearing loss in the United States. These cases are inherited in a mitochondrial pattern, which is also known as maternal inheritance. This pattern of inheritance applies to genes contained in mtDNA. Because egg cells, but not sperm cells, contribute mitochondria to the developing embryo, children can only inherit disorders resulting from mtDNA mutations from their mother. These disorders can appear in every generation of a family and can affect both males and females, but fathers do not pass traits associated with changes in mtDNA to their children.
In some cases, hearing loss occurs in people with no history of the condition in their family. These cases are described as sporadic, and the cause of the hearing loss is often unknown. When hearing loss results from environmental factors, it is not inherited.
Other Names for This Condition
- Isolated deafness
- Nonsyndromic deafness
- Nonsyndromic hearing impairment
- Nonsyndromic hearing loss and deafness
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
- DEAFNESS, AUTOSOMAL DOMINANT 7; DFNA7
- DEAFNESS, AMINOGLYCOSIDE-INDUCED
- DEAFNESS, AUTOSOMAL RECESSIVE 2; DFNB2
- DEAFNESS, AUTOSOMAL DOMINANT 4A; DFNA4A
- DEAFNESS, AUTOSOMAL RECESSIVE 9; DFNB9
- DEAFNESS, AUTOSOMAL RECESSIVE 1A; DFNB1A
- DEAFNESS, AUTOSOMAL DOMINANT 9; DFNA9
- DEAFNESS, AUTOSOMAL RECESSIVE 12; DFNB12
- DEAFNESS, AUTOSOMAL DOMINANT 12; DFNA12
- DEAFNESS, AUTOSOMAL DOMINANT 3A; DFNA3A
- DEAFNESS, AUTOSOMAL DOMINANT 2A; DFNA2A
- DEAFNESS, X-LINKED 3; DFNX3
- DEAFNESS, AUTOSOMAL DOMINANT 13; DFNA13
- DEAFNESS, AUTOSOMAL RECESSIVE 3; DFNB3
- DEAFNESS, AUTOSOMAL DOMINANT 5; DFNA5
- DEAFNESS, X-LINKED 2; DFNX2
- DEAFNESS, X-LINKED 1; DFNX1
- DEAFNESS, AUTOSOMAL DOMINANT 6; DFNA6
- DEAFNESS, X-LINKED 5, WITH PERIPHERAL NEUROPATHY; DFNX5
- DEAFNESS, AUTOSOMAL RECESSIVE 7; DFNB7
- DEAFNESS, X-LINKED 4; DFNX4
- DEAFNESS, AUTOSOMAL DOMINANT 10; DFNA10
- DEAFNESS, AUTOSOMAL DOMINANT 11; DFNA11
- DEAFNESS, X-LINKED 6; DFNX6
- DEAFNESS, AUTOSOMAL DOMINANT 20; DFNA20
- DEAFNESS, AUTOSOMAL DOMINANT 23; DFNA23
- DEAFNESS, AUTOSOMAL RECESSIVE 21; DFNB21
- DEAFNESS, AUTOSOMAL RECESSIVE 13; DFNB13
- DEAFNESS, AUTOSOMAL RECESSIVE 17; DFNB17
- DEAFNESS, AUTOSOMAL DOMINANT 15; DFNA15
- DEAFNESS, AUTOSOMAL RECESSIVE 18A; DFNB18A
- DEAFNESS, AUTOSOMAL RECESSIVE 20; DFNB20
- DEAFNESS, AUTOSOMAL RECESSIVE 16; DFNB16
- DEAFNESS, AUTOSOMAL DOMINANT 36; DFNA36
- DEAFNESS, AUTOSOMAL DOMINANT 22; DFNA22
- DEAFNESS, AUTOSOMAL RECESSIVE 31; DFNB31
- DEAFNESS, AUTOSOMAL DOMINANT 43; DFNA43
- DEAFNESS, AUTOSOMAL RECESSIVE 37; DFNB37
- DEAFNESS, AUTOSOMAL DOMINANT 48; DFNA48
- DEAFNESS, AUTOSOMAL DOMINANT 52; DFNA52
- DEAFNESS, AUTOSOMAL RECESSIVE
- DEAFNESS, AUTOSOMAL RECESSIVE 33; DFNB33
- DEAFNESS, AUTOSOMAL RECESSIVE 28; DFNB28
- DEAFNESS, AUTOSOMAL RECESSIVE 67; DFNB67
- DEAFNESS, AUTOSOMAL RECESSIVE 36, WITH OR WITHOUT VESTIBULAR INVOLVEMENT; DFNB36
Scientific Articles on PubMed
References
- Ding Y, Leng J, Fan F, Xia B, Xu P. The role of mitochondrial DNA mutations in hearing loss. Biochem Genet. 2013 Aug;51(7-8):588-602. doi: 10.1007/s10528-013-9589-6. Epub 2013 Apr 21. Citation on PubMed
- Duman D, Tekin M. Autosomal recessive nonsyndromic deafness genes: a review. Front Biosci (Landmark Ed). 2012 Jun 1;17(6):2213-36. doi: 10.2741/4046. Citation on PubMed or Free article on PubMed Central
- Hildebrand MS, Morin M, Meyer NC, Mayo F, Modamio-Hoybjor S, Mencia A, Olavarrieta L, Morales-Angulo C, Nishimura CJ, Workman H, DeLuca AP, del Castillo I, Taylor KR, Tompkins B, Goodman CW, Schrauwen I, Wesemael MV, Lachlan K, Shearer AE, Braun TA, Huygen PL, Kremer H, Van Camp G, Moreno F, Casavant TL, Smith RJ, Moreno-Pelayo MA. DFNA8/12 caused by TECTA mutations is the most identified subtype of nonsyndromic autosomal dominant hearing loss. Hum Mutat. 2011 Jul;32(7):825-34. doi: 10.1002/humu.21512. Epub 2011 Jun 7. Citation on PubMed or Free article on PubMed Central
- Hilgert N, Smith RJ, Van Camp G. Function and expression pattern of nonsyndromic deafness genes. Curr Mol Med. 2009 Jun;9(5):546-64. doi: 10.2174/156652409788488775. Citation on PubMed or Free article on PubMed Central
- Lin FR, Niparko JK, Ferrucci L. Hearing loss prevalence in the United States. Arch Intern Med. 2011 Nov 14;171(20):1851-2. doi: 10.1001/archinternmed.2011.506. No abstract available. Citation on PubMed or Free article on PubMed Central
- National Institute on Deafness and Other Communication Disorders: Quick Statistics
- Shearer AE, Hildebrand MS, Schaefer AM, Smith RJH. Genetic Hearing Loss Overview. 1999 Feb 14 [updated 2023 Sep 28]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1434/ Citation on PubMed
- Shearer AE, Hildebrand MS, Sloan CM, Smith RJ. Deafness in the genomics era. Hear Res. 2011 Dec;282(1-2):1-9. doi: 10.1016/j.heares.2011.10.001. Epub 2011 Oct 8. Citation on PubMed or Free article on PubMed Central
- Shearer AE, Smith RJ. Genetics: advances in genetic testing for deafness. Curr Opin Pediatr. 2012 Dec;24(6):679-86. doi: 10.1097/MOP.0b013e3283588f5e. Citation on PubMed or Free article on PubMed Central
- Song MH, Lee KY, Choi JY, Bok J, Kim UK. Nonsyndromic X-linked hearing loss. Front Biosci (Elite Ed). 2012 Jan 1;4(3):924-33. doi: 10.2741/E430. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.